Pemphigus Vulgaris
- Article Author:
- Curtis Ingold
- Article Editor:
- Moien AB Khan
- Updated:
- 8/10/2020 9:31:29 PM
- For CME on this topic:
- Pemphigus Vulgaris CME
- PubMed Link:
- Pemphigus Vulgaris
Introduction
Pemphigus vulgaris (PV) is an autoimmune disease that results in blisters on cutaneous and mucosal surfaces.[1] Pemphigus is derived from pemphix, the Greek word for blister. Pemphigus was first described in 1788 by Stephen Dickson, who observed a patient with a blister on her tongue.[2] Although PV has not been shown to be contagious as initially thought, there have been possible triggers identified that might induce PV in patients with other autoimmune disorders.[1]
Etiology
The etiology of pemphigus vulgaris is unknown, but patients are at risk that has a genetic predisposition. Several studies have linked PV with human leukocyte antigen (HLA) class II alleles. HLA-DRB1 0402 is associated with PV in Ashkenazi Jews, while DRB1 1401/04 and DQB1 0503 HLA alleles are associated with PV in non-Jewish patients of European or Asian descent.[3][4][5] Environmental factors in addition to diet, stress, viral infections, medications, radiation therapy, and allergens may all induce immune dysregulation leading to a flare of PV.[1]
Epidemiology
Though pemphigus vulgaris is prevalent worldwide, the occurrence of PV is related to ethnicity and geographic location. The reported incidence is between 0.1 and 0.5 per 100,000 people per year.[6] However, a higher rate is recorded in certain ethnicities.[7] Ashkenazi Jews have been found to have an increased incidence of PV, and the average onset of PV is usually seen between the ages of 40 to 60 years.[8] Also, within the demographics, people living in India, Southeast Europeans, and the Middle Eastern are at greatest risk for pemphigus Vulgaris.[9] The prevalence of PV is roughly the same in men and women. However, in Tunisia, PV is more common in women compared to men by a ratio of 4 to 1.[1][8]
Pathophysiology
Pemphigus vulgaris is caused by autoantibodies that target keratinocyte proteins (desmogleins). Acantholysis wherein there is a loss of keratinocyte to keratinocyte adhesion induced by the binding of circulating immunoglobulin G (IgG) autoantibodies to intercellular adhesion molecules.[10][11] Acantholysis is seen due to the autoantibodies destroying the intracellular connections leading to bullae that can easily rupture. A “super-compensation hypothesis” was recently submitted by Sinha et al. proposes that additional factors may also play a role in PV.[12] Multiple mechanisms for antibody-induced acantholysis have been suggested, including the induction of signal transduction and the inhibition of adhesive molecule function through steric hindrance, which can trigger cell separation.[13] Further, the pathogenesis was described in more detail by Hammers et al.[14] It has been found in patients with PV, the presence of autoantibodies against desmoglein 1 (Dsg 1) and desmoglein 3 (Dsg 3).[15] Desmogleins are transmembrane glycoproteins that are an integral part of desmosomes, which in part is required for cell-cell adhesion. Didona et al. reviewed how IgG binds desmogleins in greater detail. The most common targets on desmoglein for IgG antibodies are the extracellular cadherin domains, which can result in loss of desmosome adhesive properties, signaling pathways that trigger endocytosis and depletion, and direct inhibition of Dsg 3 trans-interactions.[16]
Murine studies have shown that enzymatic inactivation of Dsg 1 and gene deletion of Dsg 3 results in pathology that is similar to PV. Also, the addition of IgG from patients with PV to mice results in pathology that is similar to PV. This phenomenon was observed to be dose-dependent and suggests that reducing the circulating levels of IgG against Dsg 1 and Dsg 3 can improve patient outcomes. In patients with the mucocutaneous disease, it was found that they have autoantibodies against Dsg 1 and Dsg 3, whereas patients with disease localized to their mucous membranes were only found to have autoantibodies against Dsg 3. This can be explained by the desmoglein compensation model in which there are Dsg 1 and Dsg 3 in the cutaneous epidermis and that having autoantibodies against just one of the desmogleins will not cause disease. However, due to the normal lack of Dsg 1 in mucous membranes, autoantibodies against only Dsg 3 will result in disease because Dsg 1 is not present to compensate for the inhibition of Dsg 3. The binding of antibodies to desmogleins has been confirmed by epitope mapping and is presumed to disrupt desmoglein binding by affecting steric hindrance. Another proposal of the pathophysiology in PV that may occur in addition to the above explanation is the desmoglein non-assembly depletion hypothesis. This theory suggests that autoantibodies not only bind desmoglein but that they also bind each other leading to crosslinking and the inability for desmosomes to maintain cell-cell adhesion.
PV has been shown to have a genetic component, although familial cases are uncommon.[17] Patients with PV have been found to have a higher frequency of non-symptomatic first-degree relatives with circulating PV-IgG antibodies than compared to healthy controls. Additionally, first-degree relatives were found to have a higher prevalence of autoimmune diseases. DQB1*0503 and DRB1*0402 are two of the most common PV-associated alleles. DRB1*0402 was shown to be protective against rheumatoid arthritis.
Exposure to certain medications like penicillamine and captopril can trigger PV. Such a trigger can happen through the effects on binding to molecules involved in cell adhesion, influence on enzymes that mediate keratinocyte aggregation, and molecules involved in cell and by stimulating neoantigen formation.[18] Furthermore, nonsteroidal anti-inflammatory agents, penicillin, cephalosporins have also been associated with drug-induced PV.[18][19] Finally, controversial case reports associating PV with certain foods like red wine, garlic, leek, and peppers exists, though such association is not supported by robust evidence.[20]
Histopathology
The diagnosis of pemphigus vulgaris is confirmed with a biopsy of the lesion. Histopathology and Tzanck smear will reveal acantholysis. Direct immunofluorescence is considered the "gold standard" for the diagnosis of PV. Enzyme-linked immunosorbent assay (ELISA) testing will show serum IgG against Dsg 1, Dsg 3, or both in 98.5% of samples. ELISA testing for PV is commercially available.[8][21]
History and Physical
Pemphigus vulgaris is a blistering disease that initially presents on the oral mucosa in 80% of cases. These intraoral blisters often rupture, leaving painful erosions. Cutaneous lesions may appear in about 75% of patients with PV after the first oral blisters have presented. Vesicles, erosions, or bullae may appear on erythematous or normal-appearing skin. A Nikolsky sign is described as a blister formation with minor pressure or trauma and is seen in PV.[22]
Due to the painful nature of oral lesions, PV can result in an impaired nutritional status. Mucosal PV can be found in the conjunctiva, nasal mucosa, larynx, pharynx, esophagus, penis, vagina, and anus.[23] Cutaneous lesions can be found most commonly on the face, trunk, groin, scalp, and armpits. PV usually spares palms and soles. Blisters can heal without scarring but may result in changes in the pigment. Alopecia may be observed when PV affects the scalp. Rarely, PV will involve nails when the disease is severe.[8][21] Rarely PV can present as pemphigus herpetiformis where PV can present with urticarial plaques and cutaneous vesicles arranged in a herpetiform.
Evaluation
The diagnosis of pemphigus vulgaris is ascertained by obtaining a thorough history and through the use of a biopsy. The biopsy for direct immunofluorescence (DIF) should be taken from normal-appearing perilesional skin or mucosa.[24] A physical exam will most likely be positive for mucosal involvement of oropharyngeal, nasal, and genital regions. Tzanck smear and histology examination will show acantholysis.[8] Serological studies like indirect immunofluorescence (IIF) and enzyme-linked immunosorbent assay(ELISA) can detect circulating autoantibodies that bind epithelial cell surface antigens.[24]
In patients with positive DIF results, ELISA testing will show serum IgG against Dsg 1, Dsg 3, or both.[21] Additional workup should include vitals, pregnancy testing in select population, complete blood count (CBC), metabolic panel, antinuclear antibody (ANA), and urinalysis. A bone density scan may be needed early in the disease to ensure proper prevention of osteoporosis in at-risk patients. Also, quality of life and disease activity should be documented to ensure a baseline for further monitoring. Two validated severity scoring systems include the Pemphigus Disease Area Index (PDAI) and the Autoimmune Bullous Skin Disorder Intensity Score (ABSIS) that can be completed in 2 to 5 minutes.[25]
Treatment / Management
Systemic corticosteroids have had a significant impact on the treatment of pemphigus vulgaris and remain the backbone in the management of PV.[26] The first-line treatment of mild PV is systemic corticosteroids, which takes several weeks to achieve a response.[25] Tapering down of the dose can be initiated when symptoms are improved, but if reappearance of more than three lesions occurs, then dosing should be increased again to induce remission.[27]
Second-line treatment is in combination with corticosteroids and includes adding either azathioprine or mycophenolate mofetil (MMF).[27] Azathioprine is a purine analog that inhibits purine synthesis. It can be administered orally or by intravenous infusion. Azathioprine should be discontinued if no improvements are seen within three months. Dosing should be adjusted based on renal function. Close monitoring is required to reduce the likelihood of side effects. Nausea is the most common side effect seen with azathioprine. Bone marrow suppression is also seen with azathioprine that can cause pancytopenia, thrombocytopenia, and leukopenia.[28] MMF functions as an immunosuppressant by inhibiting purine synthesis. It can be administered orally or by intravenous infusion. MMF is usually effective within two months of initiating treatment. Common side effects include nausea, vomiting, diarrhea, and abdominal discomfort. Intravenous administration can cause superficial thrombophlebitis and thrombosis.[27]
Anti-CD20 monoclonal antibodies, such as rituximab and ofatumumab, have also been used in conjunction with corticosteroids for first-line treatment in moderate-to-severe pemphigus.[21] Rituximab is an anti-CD20 monoclonal antibody that stops B lymphocytes from maturing into autoantibody-producing plasma cells. It is administered intravenously, and response is usually seen within three months. Common side effects include infusion-related reactions such as nausea, vomiting, headache, and fever.[27] A rare serious side effect of rituximab is progressive multifocal leukoencephalopathy (PML) that has also been seen in other monoclonal antibodies.[29]
Third-line treatments for PV include intravenous immunoglobulin (IVIG), cyclophosphamide, dapsone, immunoadsorption, and methotrexate.[27][30]
Most of the emerging therapies that could be effective for PV are monoclonal antibodies, with the cost being a major restriction to large trials. Obinutuzumab, ofatumumab, and veltuzumab are anti-CD20 monoclonal antibodies that may be an alternative to rituximab. Other therapies that may offer hope in PV include medications that target B-cell derived B-cell activating factor (BAFF), a proliferation-inducing ligand (APRIL), CD19, Bruton kinase (BTK), and interleukin (IL-4).[16]
Future PV research should focus on better understanding the specific pathogenic molecules and cytokines, after which better therapies can be produced.[31]
Differential Diagnosis
IgA pemphigus is similar to pemphigus vulgaris in that is has painful blisters, but IgA pemphigus does not present with oral mucosa blisters. Direct and indirect immunofluorescence can both help to differentiate PV from IgA pemphigus.[32]
Pemphigus foliaceus is similar to PV in the fact that it is an autoimmune blistering disease, however similarly to IgA pemphigus, pemphigus foliaceus does not affect the oral mucosa. Pemphigus foliaceus is less common than PV.[33]
Paraneoplastic pemphigus also presents with mucocutaneous vesicles and bullae similar to PV. Paraneoplastic pemphigus can be differentiated from PV using indirect immunofluorescence and immunoblot.[34]
Prognosis
Follow up, and response to treatment of pemphigus vulgaris should be monitored closely. Pemphigus is an active disease and often requires dose adjustments and change in medications according to the response to treatment. Septicemia is the leading cause of death in PV.[8] In a retrospective cohort study by Kridin et al. in Israel, it was found that survival rates were lower in patients with PV than compared with the general population. Patients who were diagnosed with PV at an older age had a lower survival rate. The median overall survival from the point of diagnosis was 10.1 years (0.2 to 29.8 years). There was not a statistically significant survival difference between men and women.[35]
Complications
Systemic corticosteroids, which are the backbone of pemphigus vulgaris treatment, are well known for causing osteoporosis and other complications. Corticosteroids have been reported to cause fractures in 30 to 50% and cause osteonecrosis in 9 to 40% of patients receiving long-term therapy.[36] The Fracture Risk Assessment (FRAX) is a tool used to further stratify the risk of osteoporotic fracture in patients that are not osteoporotic by T score and can be valuable when considering using pharmacologic treatment for the prevention of bone loss.[37] Other common side effects from corticosteroids include hyperglycemia, insomnia, increased appetite, hypertension, edema, adrenal suppression, cataracts, and delayed wound healing.[38]
A case-control study by Namazi et al. looked at the incidence of P-wave dispersion (PWD) in patients with PV. The authors reasoned that atrial fibrillation can be predicted by the presence of PWD and hypothesized that atrial fibrillation might be higher in PV patients. Patients were excluded if they had obesity, hyperlipidemia, hypertension, diabetes mellitus, and cardiopulmonary disease. The authors stated the limitations of their study included a small sample size of 90 patients, and the use of corticosteroids was higher in the patients with PV. Regardless of those limitations, the incidence of PWD confirmed by electrocardiogram (ECG) was higher in the PV group than in the control group.[39]
Deterrence and Patient Education
Patient education should include close follow-up visits with their provider and specialists. Medication compliance is integral to the management of pemphigus vulgaris and can be re-enforced by the support of the pharmacist.
Enhancing Healthcare Team Outcomes
Pemphigus vulgaris is a life-threatening autoimmune disease for which treatment is indicated. The goal of treatment in PV is to induce complete remission of PV with minimal treatment-related adverse effects. Cutaneous and mucosal involvement in PV can cause significant pain and functional impairment. Local measures may help to improve patient symptoms. Due to the serious adverse effects, prolonged treatment with high doses of a systemic glucocorticoid is not recommended. A secondary infection like Herpes simplex should be considered when lesions fail to respond to treatment. An interprofessional approach, including social workers, wound care staff, dieticians, nursing, physician assistants, nurse practitioners, pharmacists, dentists, primary care physicians, hospitalists, and dermatologists, can optimize outcomes.
(Click Image to Enlarge)
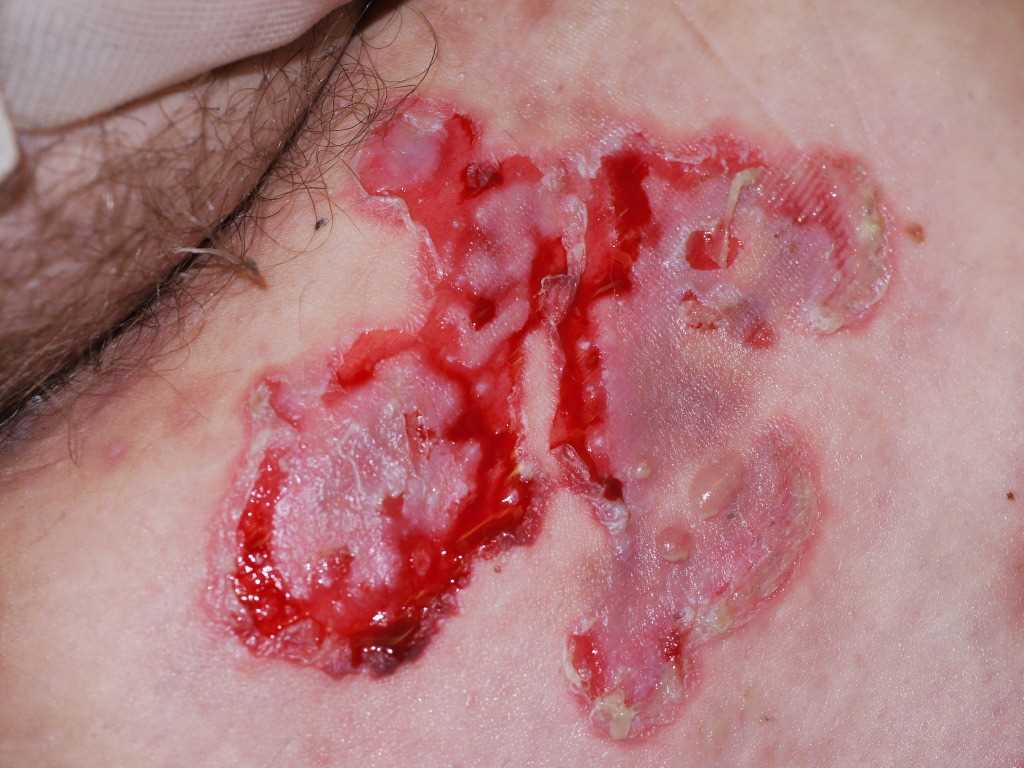
Pemphigus Vulgaris
Contributed by DermNetNZ