Polycystic Kidney Disease
- Article Author:
- Siddique Akbar
- Article Editor:
- Syed Rizwan Bokhari
- Updated:
- 8/10/2020 9:10:36 PM
- For CME on this topic:
- Polycystic Kidney Disease CME
- PubMed Link:
- Polycystic Kidney Disease
Introduction
Polycystic kidney disease (ADPKD) is an autosomal dominant disorder. It's a multisystem and progressive disease with cysts formation and kidney enlargement along with other organ involvement (e.g., liver, pancreas, spleen).
In the adult population, it is the most frequent genetic cause of renal failure, with 6% to 8% of patients on dialysis in the US. By the age of 60 years, 50% of them need renal replacement therapy. Cysts may be detected in childhood or in utero, but clinical manifestations appear in the third or fourth decade of life.
Etiology
ADPKD is an autosomal dominant disease, so it is found in males and females equally, and each offspring has a 50% chance of inheriting the disease.
ADPKD involves at least two genes. PKD1 accounts for most ADPKD cases and is located on 16p13.3. PKD2 accounts for 15% of ADPKD cases.
PKD1 codes for polycystin 1, a 4304 amino acid protein. Polycystin 1 interacts with polycystin 2 and is involved in cell cycle regulation and intracellular calcium transport. PKD2 codes for polycystin two, which is structurally similar to polycystin 1. It is a member of the family of voltage-activated calcium channels.
Polycystin 1 and two are located in the epithelial cells of the renal tubules and other areas of the renal cell epithelium. Both form heteromeric complexes and are found in the primary cilium of epithelial cells in kidneys. The primary cilium is considered a mechanical receptor that can sense changes in tubular fluid flow, and that can transduce them into an intracellular calcium signaling. ADPKD1 is more severe than ADPKD2.
Epidemiology
Clinical registry data show prevalence rates of diagnosed cases ranging from 1 in 543 to 1 in 4000. Approximately 4 to 7 million individuals are affected in the world and account for 7% to 15% of patients on renal replacement therapy. The proportion of end-stage renal disease (ESRD) caused by ADPKD is less among African Americans than among whites because of a higher incidence of other causes of ESRD
Symptoms usually increase with age. Children very rarely present with renal failure from ADPKD, and disease is slightly more severe in males.
Pathophysiology
Polycystin-1 (PKD1 protein) and polycystin-2 (PKD2 protein) belong to a subfamily of transient receptor potential (TRP) channels[1]. PKD2 is found in the endoplasmic reticulum and also in the plasma membrane in spindles in cell division and primary cilium. PKD1 inactivation determines the rate of development of the cystic disease. PC1 interacts with PC2 and helps in the maturation of PC2, and vice versa. PC1 and PC2 also interact with additional calcium channel proteins.
A common finding in animal models of PKD has increased levels of cyclic adenosine monophosphate (cAMP), not only in the kidney but also in the liver and vascular smooth muscle. cAMP exerts effects on cell proliferation in different cell types. cAMP and PKA signaling enhances several pro-proliferative pathways in cells derived from polycystic kidneys while inhibiting proliferation in cells derived from normal human kidney cortex.
Liver cysts arise by excessive proliferation and dilation of biliary ductules and peribiliary glands.
Endothelial vasodilation and constitutive nitric oxide synthase activity are reduced in subcutaneous resistance vessels from patients with ADPKD and normal glomerular filtration rate (GFR), thus causing hypertension.
History and Physical
Renal size increases with age, and renal enlargement eventually occurs in 100% of patients with ADPKD. The severity of the structural abnormality correlates with the manifestations of ADPKD, such as pain, hematuria, hypertension, and renal impairment. Most clinical manifestations are directly related to the enlargement of renal cysts. [2]
Episodes of acute renal pain are seen quite often due to cyst hemorrhage, infection, stone, and, rarely, tumors. Visible hematuria may be the initial presenting symptom[3]. Cyst hemorrhage is a frequent complication causing gross hematuria when the cyst communicates with the collecting system. It can manifest with fever, raising the possibility of cyst infection. Occasionally a hemorrhagic cyst will rupture, resulting in a retroperitoneal bleed.
Urinary tract infection (UTI) is common in ADPKD. UTI presents as cystitis, acute pyelonephritis, cyst infection, and perinephric abscesses. Most infections are caused by Escherichia coli, Klebsiella and Proteus species, and other Enterobacteriaceae.
Both CT and MRI are sensitive to detect complicated cysts.
Renal stone disease occurs in about 20% of patients with ADPKD. Most stones are composed of uric acid, calcium oxalate, or both. Stones can be difficult to diagnose on imaging in ADPKD because of cyst wall and parenchymal calcification. CT urography is helpful.
Hypertension is the most common manifestation of ADPKD. Microalbuminuria, proteinuria, and hematuria are more common in hypertensive patients with ADPKD.
In a majority of patients, renal function is within the normal range. By the time renal function starts worsening, the kidneys usually are greatly enlarged. ESRD is not inevitable in ADPKD. Up to 77% of patients are alive with preserved renal function at age 50 years, and 52% at age 73. Kidney and cyst volumes are the strongest predictors of renal functional decline.
Most simple hepatic cysts are solitary, and PLD should be suspected when four or more cysts are present in the hepatic parenchyma.
Evaluation
Ultrasound criteria for the diagnosis of ADPKD:
Original Ravine’s PKD1 Diagnostic Criteria[4]
- Age 15-29 years - two or more cysts unilateral or bilateral
- Age 30-39 years - two or more cysts in each kidney
- Age 40-49 years - two or more cysts in each kidney
- Age 60 years or above - four or more cysts in each kidney
Genetic testing is done when a precise diagnosis is not confirmed enlargement along, and the results of imaging are indeterminate.
Treatment / Management
Flank Pain:
Causes of flank pain that may require intervention, such as infection, stone, and tumor, should be excluded. Opioid analgesics should be reserved for the management of acute pain. Reassurance, lifestyle modification, and avoidance of aggravating activities may be helpful. Tricyclic antidepressants are helpful as in other chronic pain syndromes, as they are well tolerated. Cyst aspiration, under ultrasound or CT guidance, can be done if distortion of the kidney by large cyst is considered the cause of the pain. If multiple cysts are contributing to pain, laparoscopic or surgical cyst fenestration may be of benefit.
Cyst hemorrhage:
Cyst hemorrhage episodes are self-limited, and patients respond well to conservative management with bed rest, analgesics, and increased fluid intake to prevent obstructing clots. Rarely, bleeding is more severe, leading to hemodynamic instability; this requires hospitalization and transfusions.
Cyst and urinary tract infection:
Immediate treatment of symptomatic cystitis and asymptomatic bacteriuria is indicated to prevent retrograde seeding of the renal parenchyma[5]. Agents of choice include trimethoprim-sulfamethoxazole and fluoroquinolones. If fever persists after 1 to 2 weeks of appropriate antimicrobial therapy, infected cysts should be drained percutaneously or surgically. In the case of end-stage polycystic kidneys, nephrectomy should be considered.
Nephrolithiasis:
Potassium citrate is the treatment of choice in stone-forming conditions associated with it, which are uric acid stones, hypo-citraturic calcium oxalate stones[3], and distal acidification disorders.
Hypertension:
Angiotensin-converting enzyme (ACE) inhibitors or angiotensin receptor blockers (ARBs)[6] increase renal blood flow in ADPKD and are antihypertensives of choice.
Differential Diagnosis
- Multiple benign simple cysts
- Localized renal cystic disease
- Acquired renal cystic disease
- Medullary sponge kidney
- Bilateral parapelvic cysts
- Autosomal recessive polycystic kidney disease
- Tuberous sclerosis complex
- Von Hippel-Lindau syndrome
- Autosomal dominant medullary cystic disease
- Autosomal dominant polycystic liver disease
- Orofaciodigital syndrome type I
- Bardet-Biedl syndrome
- Renal cysts and diabetes syndrome
Complications
- End-stage renal disease
- Hypertension
- Polycystic liver disease
- Cerebral aneurysms
- Renal stones
- Infections
Enhancing Healthcare Team Outcomes
Polycystic kidney disease is a systemic disorder that affects many organs, and hence an interprofessional approach to management is necessary. A nephrologist, surgeon, invasive radiologist, neurosurgeon, cardiologist, and a nephrology or dialysis nurse are key professionals required to manage these patients. The pharmacist needs to educate the patient on blood pressure control and undergo regular blood testing to assess renal function. Patients need to be educated about the complications that may include cerebral aneurysms, kidney stones, and end-stage renal disease.[7][8]
The PRO-PKD score has been developed to predict the prognosis of ADPKD. As uncontrolled hypertension accelerates the decline in renal function, it is essential to control the blood pressure properly. Pain control and early treatment of cyst infection or urinary tract infection can improve the quality of life in ADPKD patients. The education of patients about the condition can reduce the number of hospitalizations. [9][10](Level 5)
(Click Image to Enlarge)
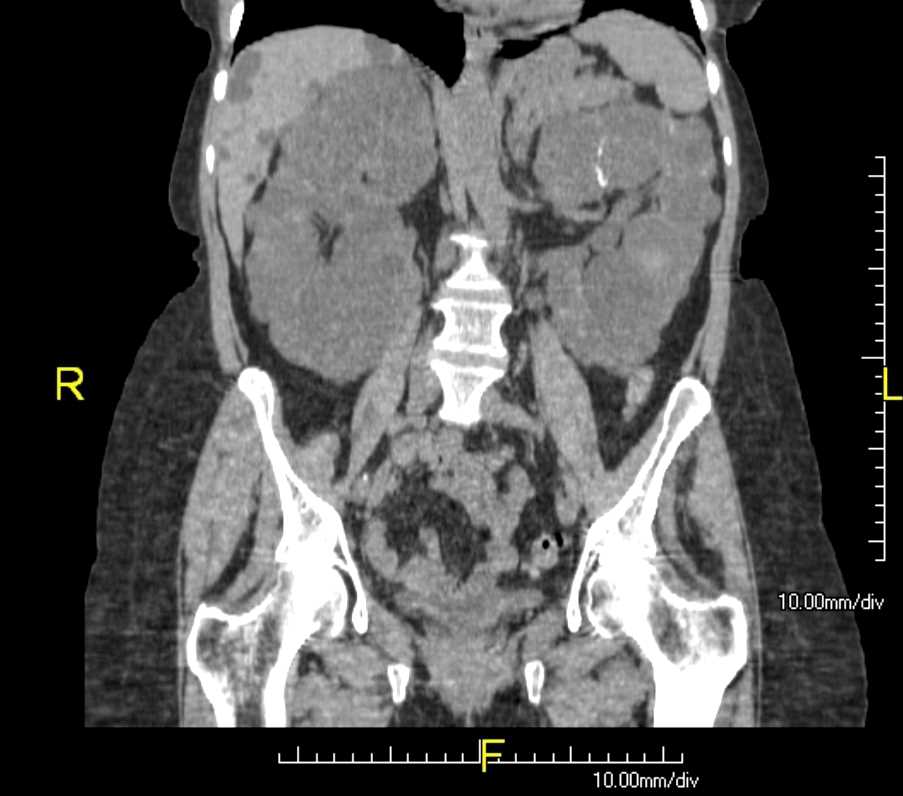
Polycystic Kidneys (adpkd) and Liver Cysts CT Abdomen Coronal view
Contributed by Scott Dulebohn, MD
(Click Image to Enlarge)

Polycystic Kidney
Contributed by Dr. Michael Lambert
(Click Image to Enlarge)
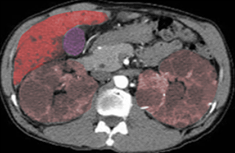
AutosomalPolycystickidney
Image courtesy S Bhimji MD