Retinitis Pigmentosa
- Article Author:
- Teri O'Neal
- Article Editor:
- Euil Luther
- Updated:
- 8/10/2020 9:37:45 PM
- For CME on this topic:
- Retinitis Pigmentosa CME
- PubMed Link:
- Retinitis Pigmentosa
Introduction
Retinitis pigmentosa as a clinical entity was originally described in 1853, but the name was not attached to the disease until 1857.[1] Considered by most to be a misnomer, the term retinitis persists today, even though inflammation has only a small role in the natural progression of the disease. Retinitis pigmentosa (RP) is not a single entity, but rather a group of disorders which produce a gradual loss of vision. Also known as hereditary retinal dystrophy, it affects 1 in every 4000 people in the United States, and approximately one in 5000 worldwide, making RP the most common inherited disease of the retina. It is usually bilateral, but there have been reports of unilateral eye involvement with RP. While it may present and progress with a variety of clinical manifestations, the first symptom of RP is generally nyctalopia, or loss of night vision, which is followed by a gradual narrowing of the visual fields. Over time, depending on the severity and rate of progression of the disease, tunnel vision or complete vision loss can be the result. As the disease progresses, other features may develop as well, including loss of accurate color discrimination, and eventual loss of visual acuity. Most patients will maintain some light perception even with late-stage RP, as the macula continues to function. Perhaps the most unsettling late effect of RP is the development of photopsia (perceived flashes of light), likely due to sensory deprivation. This may even progress to the point of visual hallucinations.
The disease may involve vision loss alone, and in this event is referred to as "nonsyndromic" RP. The majority of RP cases, about 70% to 80%, fall into the nonsyndromic category. When RP occurs in conjunction with systemic disease, it is termed "syndromic" RP. The most common form of syndromic RP is Usher syndrome, which involves a neurosensory hearing loss in association with vision loss.[2]
Etiology
Genetic mutations responsible for retinitis pigmentosa produce biochemical dysfunction specifically affecting rod photoreceptors in the retina. Defects may be associated with multiple pathways of injury including apoptosis, light damage, ciliary transport dysfunction, and endoplasmic reticulum stress. The common result of all the possible pathways is the death of the rod photoreceptors.[3] Since the rods are responsible for low-light vision, the ever-increasing loss of these cells produces the characteristic night blindness associated with RP and a gradual diminution of peripheral vision. Eventually, the destruction of large numbers of rods has a deleterious effect on the retinal pigment epithelium (RPE) and begins to affect cone photoreceptors as well. As cones begin to succumb to the toxic environment created by progressive cell death in the retina, dyschromatopsia, or disturbance of color perception, may develop.
More than 100 genetic loci on 50 different genes have been found to cause multiple patterns of inheritance and expression for retinitis pigmentosa. Approximately 20% of RP cases are autosomal recessive, with 10% to 20% autosomal dominant, and 10% X-linked recessive. The remaining cases are termed sporadic, and no family history or known molecular basis is found.
Epidemiology
Nonsyndromic retinitis pigmentosa has a worldwide prevalence of about one in 5000, and carriers number about one in 1000. Males are affected slightly more often than females due to the X-linked form expressing more frequently in males. Syndromic RP is much less common, with estimates for Usher syndrome ranging from 1.8 to 6.2 cases per 100,000 individuals. The average age of symptom onset is dependent on the genetic type. The autosomal recessive form will develop symptoms in the early teen years, but those affected with autosomal dominant RP will likely not have symptoms until well into their 20s. More than three-quarters of individuals with RP will be symptomatic and present for clinical evaluation and diagnosis of the disease by the time they are 30 years of age.
Pathophysiology
As previously mentioned, there are multiple genetically directed mechanisms for the progress of retinitis pigmentosa. Apoptosis is essentially physiologic programming for cell death, which can be triggered by a genetic mutation. Apoptosis can also be induced through cell to cell communication between the photoreceptors themselves.[4] Hence, the death of rods can eventually spread to the cone receptors as well. Light exposure may exacerbate phototoxic mechanisms. These include mutations in retinol metabolism, and acceleration of oxygen consumption in the environment, which can enhance the degeneration of photoreceptors, both rod, and cone.[5] The ciliary function is important to the transport of nutrients and other substances in the retina. Some genetic mutations, including the one for Usher syndrome, can impair this function and cause cell vulnerability.[6] Stress in the endoplasmic reticulum can cause the release of free radicals, with subsequent stimulation of hypoperfusion of the retina and vascular endothelial cell damage.[7]
Three clinical findings typical of retinitis pigmentosa are the presence of bone spicule pigmentation, vascular narrowing, and optic nerve pallor. The melanin pigment deposits, named for their characteristic bone spicule star shape, are due to retinal pigment epithelial cells which detach and migrate to perivascular locations in the retina. The exact cause of this migration is not fully understood, nor is the narrowing of retinal vessels, although one suggestion is that this results from a decreased metabolic demand due to the death of large numbers of photoreceptors. Change in the appearance of the optic disc is probably due to the formation of glial cells which cover the disc and increase reflectivity, producing a"waxy pallor."[1]
History and Physical
The typical presentation of RP involves complaints of visual disturbances beginning early in life. Usually, this includes a relatively vague sense of being unable to see well with low light situations, or those requiring rapid adaptation from light to dim environments. Some individuals note difficulty driving at night, as oncoming headlights and other sources of bright light in the environment make it difficult for them to readjust to the darkness afterward. Narrowing of the visual fields is not initially obvious but will become apparent over time.
A genetic pedigree for the patient's family will be critical to determine the type of inheritance pattern and assist with prognosis. A complete history and review of systems are needed to identify other systems affected in addition to the visual impairment to identify syndromic variants of RP. Additionally, a review of possible exposure to infectious disease or toxins that might produce a "mimic" of the disease, should be undertaken.
Physical findings include the "classic triad" seen on a fundoscopic exam of bony spicule pigmentation, vascular narrowing, and the abnormal pallor of the optic disc. These may not be evident early in the disease, and the degree to which abnormalities are seen is variable with the severity of the disease. Other associated physical findings may include subcapsular cataracts and macular edema. While the external examination of the eye is usually normal, RP patients are at higher risk for keratoconus compared to the general population. Still, the development of keratoconus is quite rare.[8] Refractive errors also occur more commonly in RP patients than in the general population.[1]
Evaluation
Complete ophthalmologic evaluation for any individual suspected of RP is needed, including expert fundoscopic examination and the evaluation of retinal function. These tests will establish a baseline and assist in determining the rate of progression and formulating a prognosis for the disease. Visual field assessment with kinetic perimetry is considered the most effective way to evaluate the loss of peripheral vision. Color vision assessment will be needed to determine the presence and progression of dyschromatopsia, indicating the degree of cone photoreceptor involvement. Electrophysiologic evaluation of the entire retina is also helpful, as electroretinographic abnormalities may be detected early, even before nyctalopia and fundoscopic abnormalities are present.[1]
Retinal imaging has made significant progress. In the past, fluorescein angiography was the predominant method employed, but newer, non-invasive modalities have made studies easier to perform and more sensitive in detecting abnormalities. Adaptive optics scanning laser ophthalmoscopy (AOSLO) enables high-resolution evaluation of the retina to detect photoreceptor damage early in the disease. This process is likely to be of use in monitoring the progression of the disease as well as evaluating the effectiveness of treatment efficacy.[1]
No specific laboratory testing is indicated, unless there is suspicion for other disease processes contributing to vision loss, such as syphilis or cytomegalovirus (CMV) retinopathy, or unless confirmatory genetic testing is indicated.
Treatment / Management
There are no standard treatments for patients with retinitis pigmentosa. The most widely recommended treatment for many years has been supplementation with vitamin A, which some studies have shown to slow the rate of retinal deterioration.[8] However, a recent Cochrane review found no significant benefit to vitamin A for RP.[9] When individual patients are supplemented with high dose vitamin A, liver function tests should be monitored.
In recent years genetic causes of RP have been better understood, and novel treatments are being developed to combat the disease. Gene-specific or mutation-specific investigations point to the possibility that gene augmentation therapy might be designed to restore normal gene expression in photoreceptors. Other research involves cell replacement therapy, which involves transplanting retinal progenitor cells (or non-ocular stem cells) into the eye, to repopulate the retina with functional photoreceptors.
Multiple types of electronic retinal implants exist and have shown great promise in restoring partial vision in patients with end-stage disease. A kind of technology that has been of interest uses auditory information to substitute for visual sensory input.[10] While these avenues are very promising for vision restoration and preservation, there are complicated issues for rehabilitation management of these patients, as well as device-specific challenges, such as functional longevity.[1]
Differential Diagnosis
The differential diagnosis for progressive vision loss is complex. Retinitis pigmentosa is a clinical diagnosis that may be confirmed by the presence of bilateral eye involvement with night vision disturbance and gradual loss of peripheral vision. Physical findings on fundoscopic examination reveal bone spicule pigmentation, with vascular narrowing and optic disc pallor. It should also be noted that there are variants of RP with significant differences in physical findings. Retinitis Punctata Albescens (RPA) frequently presents as night blindness in childhood. On direct fundoscopic examination, findings include less prominent (or even absent) disc pallor, less pronounced vascular narrowing, and bony spicules are rare or absent altogether. Instead, there are small white spots covering the majority of the fundus in RPA. Awareness of such variants is necessary to avoid excluding RP and its subclasses from the differential.[11]
Other diseases may mimic the early findings, and careful evaluation must be undertaken to make the correct diagnosis. One possible mimic is cone/rod dystrophy. This progressive retinopathy can be differentiated from RP by the onset of dyschromatopsia before nyctalopia, as the cone photoreceptors are affected rather than the rods. Another consideration for nyctalopia in childhood is Congenital Stationary Night Blindness (CSNB). The impairment in low light vision is present at birth and unlike RP, is generally non-progressive. [12][13]
Chorioretinal infections, such as syphilis, CMV or even Lyme disease may produce symptoms suggestive of RP. A careful history will usually help identify these possibilities, and laboratory testing for confirmation will be indicated.
Vision loss can be a result of multiple other diseases, including sarcoidosis and systemic lupus erythematosus. Systemic involvement in addition to vision loss should prompt a thorough assessment of these and other inflammatory processes. Vitamin A deficiency and trauma should also be considered.
Prognosis
The prognosis for patients with retinitis pigmentosa is dependent on the age of onset, and pattern of inheritance. Early-onset symptoms and severe vision loss and night blindness are expected with the autosomal recessive form of RP. The autosomal dominant expression is the least severe and associated with the more gradual onset of symptoms later in adult life. The most severe vision loss occurs with X-linked recessive RP. Tunnel vision is expected late in the course of all forms of RP, and almost all RP patients will be legally blind at some point in the progression of their disease. Total loss of vision is fortunately uncommon, as the macular function will generally allow light perception, even after acuity is lost.
Deterrence and Patient Education
Once the diagnosis of retinitis pigmentosa is made, patient and family education becomes a primary goal. Understanding the type of disease which affects the patient is very important. In a study involving vision-related quality of life and coping mechanisms for managing RP, results indicated that the large majority of individuals with RP were unaware of their subtype. Without this understanding, it is obvious that these patients cannot fully understand their disease progression and implications for their future. [14]
While no treatment options guarantee to arrest the progression of vision loss, patients should be made aware of the possible benefits of several interventions. Nutritional supplementation, especially with vitamin A, is still an area of interest, though no definite benefits are identified. Avoidance of excessive bright light exposure has been a recommendation for many years, as it may decrease phototoxic effects on the retina. Good sunglasses are therefore a must.
Patient education should also include preparation for the anticipated progression of RP. Once the diagnosis is made and the prognosis becomes relatively predictable, the patient and family must look toward adaptive changes to enable the patient to function to maximal independence for as long as possible. This involves things as simple as not rearranging the furniture in the home and installing additional lighting in work areas. As vision impairs the ability to drive and function outside the home without assistance, referral to community support agencies should be considered. As the disease affects independence and the patient begins to feel isolated, depression can be a serious complication. Patients and family members should be educated about the warning signs of depression, and clinicians should maintain vigilance in assessing the mental health of their patients.
Awareness of ongoing research and development of new therapies should be promoted as well. This knowledge will enhance the patient's ability to make an informed decision about treatment options when they become available in the future. The National Eye Institute of the National Institutes of Health is an excellent source for information, as are the Retinitis Pigmentosa Foundation Fighting Blindness and the American Foundation for the Blind. These organizations can put patients and their families in touch with support groups and other sources for assistance as well as providing information about the disease.
Pearls and Other Issues
Electroretinography (ERG) not only assist in the diagnosis of Retinitis Pigmentosa (RP) but can quantify severity and track progression of the disease. Over time, a waves decrease in amplitude to a point at which the ERG signal can no longer be detected at all. [1]
Congenital Stationary Night Blindness (CSNB), which presents with nyctalopia in childhood, presents in two forms. The first is CNSB (both Riggs and Schubert-Bornstein type) and lacks any abnormalities on the fundoscopic exam. The second group has fundoscopic abnormalities and includes both Oguchi Disease and Fundus Albipunctatis. ERG in these diseases generally demonstrates attenuation or absence of b waves, with relative preservation of a waves. As previously mentioned, these are non-progressive in nature. [13]
Enhancing Healthcare Team Outcomes
The evaluation and treatment of retinitis pigmentosa require educating the patient and family. While the disorder is primarily managed by the ophthalmologist, an ancillary team consisting of a geneticist and pharmacist may also help. Genetic testing and counseling should be considered for those affected by RP. The long-term prognosis can be better predicted in many cases, and the possibility of gene transmission to descendants will be an important consideration for some patients.
While no treatment options guarantee to arrest the progression of vision loss, patients should be made aware of the possible benefits of several interventions. Nutritional supplementation, especially with vitamin A, is still an area of interest, but the pharmacist should warn the patient that no definite benefits have been identified. In addition, Vitamin A intake for the long term has to be monitored because of its potential toxicity to the liver. The ophthalmology nurse should educate the patient on avoidance of excessive bright light exposure, as it may decrease phototoxic effects on the retina. Good sunglasses are therefore a must.
Understanding the type of disease which affects the patient is also very important. Without this understanding, patients cannot fully appreciate and learn to adapt to their disease progression and its implications. An interprofessional approach involving a specialty-trained nurse and clinician assisting in patient management and education will provide the best outcome. [Level 5]
(Click Image to Enlarge)
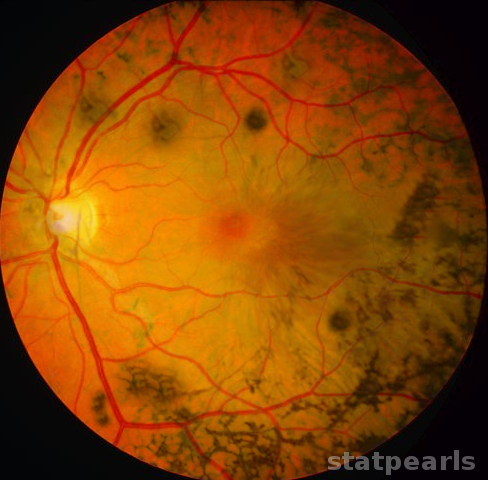
Retinitis pigmentosa
Image courtesy S Bhimji MD