Sarcoidosis
- Article Author:
- Syed Rizwan Bokhari
- Article Author:
- Hassam Zulfiqar
- Article Editor:
- Abeera Mansur
- Updated:
- 9/8/2020 8:47:28 AM
- For CME on this topic:
- Sarcoidosis CME
- PubMed Link:
- Sarcoidosis
Introduction
Sarcoidosis is a multisystem disorder of unknown etiology that mostly affect young adults worldwide and presents with noncaseating granulomas in various organs. Characteristically it presents with bilateral hilar lymphadenopathy and reticular opacities in the lungs. Other major involved sites include skin, eyes, and joints, although it can express to a variable degree in the musculoskeletal system, reticuloendothelial system, exocrine glands, heart, kidney, and central nervous system.[1][2][3]
Etiology
It is an inflammatory disease of unknown etiology that manifests as noncaseating granulomas in multiple systems. Various associations have been described, including occupational and environmental exposures to beryllium, dust, and other agents causing asthma. Various microorganisms like mycobacteria and propionibacteria have been associated. Possible infective etiology has been described in a few studies where sarcoidosis developed in a previously negative individual after cardiac or bone marrow transplantation.[4][5]
Genetic component and disease in more than one family member are usually related to antigens of the major histocompatibility complex (MHC), especially DR alleles. Few studies have described other less common genomes and angiotensin-converting enzyme genotypes in a few patients.
Cytokines including Th1, IL-2, IL6, IL 8, IL12, IL 18, IL 27, and interferon (IFN) gamma, and tumor necrosis factor (TNF) alpha are closely associated with sarcoidosis. Few of these are implicated in granuloma formation with macrophage and epithelioid accumulation, activation, and aggregation. Some of these Interleukins are believed to act as disease modifiers.
Epidemiology
The incidence is 11 cases per 100,000 population in whites but 34 cases per 100,000 population in African Americans with a lifetime risk of 2.4 percent in the USA. Extrapulmonary sarcoid is seen in up to 25 to 30 percent of patients. Cardiac involvement is seen more commonly in males while skin and eye features are more prominent in women. Extrapulmonary features can be different in terms of age of presentation, gender, and ethnicity.[6][7]
Pathophysiology
Pathogenesis of cutaneous sarcoidosis is poorly understood and attributable to both genetic and environmental factors. Key role in the development of sarcoidosis is played by T cells as they promote cellular immune reaction and usually associated with an inverted CD4/CD8 ratio. It is well characterized by noncaseating granuloma typically containing macrophages, multinucleated giant cells and epithelioid cells, in addition to lymphocytes, monocytes, mast cells, and fibroblasts There is a role of tumor necrosis factor (TNF) and TNF receptors, both are increased in this disease. This has been explained by the role of anti-TNF agents, such as pentoxifylline and infliximab. In addition to T cells role, B cell hyperreactivity with immunoglobulin production is implicated as well. Active sarcoidosis has also been associated with plasmatic hypergammaglobulinemia. Angiotensin-converting enzyme (ACE) levels correlated with elevated Soluble HLA class I antigens levels in serum.[8]
Histopathology
Biopsy of lymph nodes will reveal non-caseating granulomas.
History and Physical
Symptoms are variable; typically patients present with a persistent dry cough, fatigue, and shortness of breath. Other symptoms include painful red lumps on the skin, uveitis with the blurring of vision, hoarseness of voice, palpable lymph nodes at multiple sites including armpit, neck, painful swollen joints, hearing loss, seizures, or psychiatric disorders could be observed as part of neurological manifestations. Cardiomyopathy, conduction defects, renal calculi, and enlarged liver are observed in a few cases.
A wide range of cutaneous manifestations can be classified as papular, maculopapular, nodular, subcutaneous, hypopigmented and plaque sarcoidosis. Most common lesions in cutaneous sarcoidosis are Papular sarcoidosis involving the upper half of face, back of the neck and previous trauma /scar sites and tattoos A variant of cutaneous sarcoidosis that presents with violaceous or erythematous papules, plaques, or nodules mainly involving the central facial skin is called lupus pernio. Other well-described skin manifestations of sarcoidosis include nodular sarcoidosis. Plaque-like lesions and subcutaneous non-tender nodules are also commonly seen.
Erythema nodosum (EN) is seen in a variety of other conditions including sarcoidosis and usually presents with painful nodules on shins. It is characterized as panniculitis and is a part of Lofgren syndrome. Skin lesions can appear up to 10 or more years after the initial injury or tattoo.
Ocular manifestations are seen in close to 50% of patients of which the most common clinical feature is uveitis. The CD$/CD8 ratio of vitreous lymphocytes has prognostic value.
Heart block and sudden death have also been reported in sarcoidosis. Prophylactic insertion of ICD is recommended in patients who develop cardiac sarcoidosis.
CNS manifestations include diabetes insipidus followed by hyperprolactinemia.
The quality of life of symptomatic patients is poor. Many develop psychiatric symptoms including depression and anxiety.
Evaluation
Lab tests:
- Complete blood count and differential looking for anemia, leukopenia, and thrombocytopenia, liver function tests, blood urea nitrogen, creatinine, glucose, electrolytes, serum calcium looking for hypercalcemia. ESR and C-reactive protein are nonspecific tests, often elevated.
- Elevated serum alkaline phosphatase concentration suggests diffuse granulomatous hepatic involvement.
- Serologic tests including serum angiotensin-converting enzyme (ACE), adenosine deaminase, serum amyloid A, and soluble interleukin-2 receptor can be considered.
- Kveim test is similar to tuberculin skin test and evokes a sarcoid granulomatous response. It is of limited significance.
Radiographic tests:
- Lungs are the main site of involvement; imaging tests include chest radiograph, CT Chest, fluorine-18-fluorodeoxyglucose-positron emission tomography (FDG-PET), gallium-67, thallium-201, technetium sestamibi (MIBI-Tc) and single-photon emission computed tomography (SPECT).
- Cardiac or CNS affected sarcoidosis is better diagnosed with the help of an MRI or PET scan.
- Pulmonary function tests may reveal a decrease in DLCO and a restrictive pattern is seen in advanced cases. About 10% of patients will have an obstructive pattern. Patients with DLCO less than 60% predicted combined with oxygen saturation of less than 90 on the walk test need to be evaluated for pulmonary hypertension.
Radiographic stages are as follows:
- Stage I: Presence of bilateral hilar adenopathy
- Stage II: Bilateral hilar adenopathy and reticular opacities
- Stage III: Reticular opacities with shrinking hilar nodes ( mainly infiltrates)
- Stage IV: Reticular opacities with Fibrosis
In the case of clear chest X-ray in a patient with unexplained dyspnea or cough, (HRCT) of the chest should be considered.
Histopathology: characteristically noncaseating granulomas, with aggregates of epithelioid histiocytes, giant cells, and mature macrophages are seen.
In order to rule out systemic disease, pulmonary function test, electrocardiogram, echocardiography, urinalysis and tuberculin skin testing should be considered.
Follow-up of the pulmonary disease can be done with pulmonary function tests and a carbon monoxide diffusion capacity test of the lungs for carbon monoxide (DLCO).[9][10][11]
A biopsy is often required to confirm the diagnosis. Transbronchial biopsy has a high yield. If that fails, then a mediastinoscopy to perform a lymph node biopsy is required. The key feature is noncaseating granulomas in the absence of mycobacteria and fungi.
Treatment / Management
Pulmonary sarcoidosis is often asymptomatic, non-progressive disease and requires no treatment, as a majority of patients undergo spontaneous remission. Close monitoring of symptoms, chest radiograph, and pulmonary function is continued at three to six-month intervals should be considered in asymptomatic patients. Patients with pulmonary sarcoidosis causing worsening symptoms, stage II-III radiographic findings should be considered for oral glucocorticoids at 0.3 to 0.6 mg/kg for 4 to 6 weeks. If there is no improvement in symptoms, radiographic abnormalities, and pulmonary function tests, steroids may be continued for an additional four to six weeks. Maintainance steroids are not needed, steroid tapering to a dose of 0.25 to 0.5mg/kg (usually 10 to 20 mg) per day, should be considered over a period of at least six to eight months. Methotrexate, azathioprine, infliximab, leflunomide, and antimalarial agents may be considered as steroid-sparing agents in patients who are unable to tolerate steroids.[12][13]
Lung transplant is an option for patients with end-stage lung disease. However, the transplant is also associated with risks and the need to take immunosuppressive therapy for life.
Differential Diagnosis
- Tuberculosis
- Cat scratch disease
- Lung cancer
- Lymphoma
- Occupational lung disease
- Fungal infection
Prognosis
Asymptomatic patients do not require treatment and often remain stable for many years. However, those who develop symptomatic lung or extrapulmonary disease tend to have a guarded prognosis. Relapse of symptoms is common and many patients with advanced disease develop dyspnea and pulmonary hypertension. The overall mortality rate for untreated patients is about 5%. However, prolonged treatment with corticosteroids is not benign either and the adverse effects of these drugs are common.
Complications
Pulmonary hypertension
End stage lung disease
Enhancing Healthcare Team Outcomes
Sarcoidosis has no obvious cause and hence prevention is not possible. The disorder also resolves spontaneously and hence treatment is not always required. However, severe cases do need to follow up. The management of patients with sarcoidosis is best done in an interprofessional fashion with a team of healthcare workers that includes a cardiologist, neurologist, radiologist, pulmonologist, cardiac nurse, respiratory therapist, and a pharmacist. The patient needs regular chest x-rays since it is a marker for disease progression. Some patients may benefit from pulmonary rehabilitation and the use of bronchodilators. If the disease is advanced, a regular eye exam is important.
Symptomatic patients need drug therapy and the pharmacist needs to emphasize the importance of compliance and the need to follow up since most drugs have adverse side effects. Patients who fail to respond to steroids may need potent biological agents. In addition, patients with end-stage lung disease may need to be seen by a transplant nurse to determine their eligibility. Further patients with eye symptoms should be referred to an ophthalmologist.
At each clinic visit, the nurse should ensure that the patient gets a regular 12-lead ECG because the heart block is not uncommon. The pharmacist and nurse should educate the patient on the importance of discontinuing smoking and abstaining from alcohol. Finally, patients who are managed with steroids should be educated by all members of the team about the side effects of the drugs used. Only through such an interprofessional team approach can the outcomes of sarcoidosis be improved. [Level 5][14][15](Level V)
Outcomes
In many patients with sarcoidosis, bo treatment is required and the disorder spontaneously resolves. However, in a certain number of people, the disease may take a fulminant course with severe symptoms. Factors that indicate a poor prognosis include significant chest imaging findings, extrapulmonary involvement, and presence of pulmonary hypertension. Many studies indicate that a chest x-ray is an excellent marker for disease prognosis. In severe cases, patients may require oxygen, experience heart block and respiratory failure. Data regarding mortality are not available because in many cases long-term follow up is missing. Overall, it appears that about one-fifth of patients develop functional impairment and there is a mortality rate of 3-5% in patients who are not treated. The highest mortality rates are in African American females past the fifth decade of life. [16][17](Level V)
(Click Image to Enlarge)
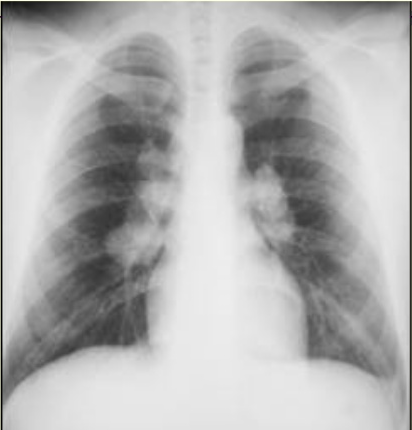
Bilateral hilar adenopathy
Contributed by Scott Dulebohn, MD