Sickle Cell Anemia
- Article Author:
- Ankit Mangla
- Article Author:
- Moavia Ehsan
- Article Editor:
- Smita Maruvada
- Updated:
- 10/7/2020 2:22:38 PM
- For CME on this topic:
- Sickle Cell Anemia CME
- PubMed Link:
- Sickle Cell Anemia
Introduction
Sickle cell anemia is the most severe form of sickle cell disease and is the homozygous state for hemoglobin S. Sickle cell anemia is prevalent in Africa, the Middle East, and parts of India. It is common in geographical areas where malaria is widespread. Hemoglobin in most individuals is present in soluble form. However, in sickle cell disease, hemoglobin precipitates as insoluble crystals which lead to an abnormal shape and size of RBCs with subsequent phagocytosis of the effected corpuscles.[1],[2],[3]
The disorder typically affects Africans and people of Mediterranean ancestry. Carriers of the sickle cell trait do not suffer from the disease.
Etiology
A point mutation in the beta globin chain of the hemoglobin causes sickle cell disease. Specifically, it occurs when a single base from A to T in the codon for glutamic acid at position 6 is changed to valine of the beta globin. If this mutation affects both of the beta globin chains, sickle cell anemia occurs; if only one chain is affected, it results in the sickle cell trait.[4]
The vaso-occlusive crises are triggered by:
- Hypoxia
- Cold weather
- Infection
- Acidosis
- Dehydration
- Alcohol
- Stress
- Pregnancy
Aplastic crises may be precipitated by:
- Folic acid deficiency
- Parvovirus B10 infection
- Ingestant of toxins like phenylbutazone
Epidemiology
This disease is predominantly present in individuals of African origin but also affects people of Middle Eastern, Indian, and Mediterranean descent. It is estimated that 1 in 13 children of African descent suffers from the sickle cell trait. Sickle cell disease affects 1 in 365 individuals of African descent; in America, about 100,000 individuals are currently suffering from this disease.[5]
Pathophysiology
A point mutation in the beta globin chain gene, where T replaces A which changes the codon for glutamic acid to valine, causes sickle cell anemia. This mutation's presence in both chains causes a change in hemoglobin. Normal hemoglobin is soluble, and it does not precipitate in the presence of hypoxia, low pH, and dehydration. The polymerization of the sickle cell hemoglobin molecules inside the RBCs is responsible for the sickling. This polymerization with subsequent aggregation takes place when the sickle cell hemoglobin is deoxygenated. Initial aggregation and polymerization is a reversible process. It occurs while the hemoglobin is deoxygenated and reverses with oxygenation. The sickle or holly leaf shape of the RBCs conforms to the shape of the polymerized hemoglobin. However, multiple cycles of sickling cause damage to the RBCs' cell membranes, making them prone to phagocytosis by the macrophages. This phagocytosis leads to the destruction and reduction of the RBC count and hence results in anemia. Reversibly sickled cells revert to normal shape with oxygenation and continue to perform their functions, but when the hemoglobin is irreversibly polymerized oxygenation cannot reverse the pathology. [6]
The deformed sickle cells have a strong adherence to the endothelium, leading to vaso-occlusive crisis.
Histopathology
The RBCs with polymerized hemoglobin appear as elongated sickles or holly leaves when observed under a microscope. The normal RBCs may have basophilic DNA particles because of functional asplenia called Howell-Jolly bodies. The cell membrane of the sickled cells is thick and usually damaged. Some of the RBCs may have a mean corpuscular volume greater than 100 femtoliters due to deficiency of folic acid because of enhanced hematopoiesis. On air-dried slides, the sickled cells are elongated with both ends pointed.
History and Physical
Sickle cell disease usually manifests after six months of age when the fetal hemoglobin levels begin to fall. This timing occurs because fetal hemoglobin keeps the sickle cell hemoglobin in solubilized form. The most common presenting feature is vaso-occlusive crises. These vaso-occlusive crises may present in a variety of ways. Patients commonly complain of excruciating pain in the abdomen, thorax, joints, long bones, and digits. Some individuals may experience multiple episodes while others may remain free of them for long periods of time. Signs and symptoms of anemia are also prevalent, including palpitations, fatigue, pallor, and tachycardia. Repeated vaso-occlusive crises may result in splenic infarctions and resultant functional asplenia. This asplenia results in repeated infections with encapsulated bacteria like Streptococcus pneumoniae, Staphylococcus aureus, and many others. These pathogens may cause life-threatening pneumonia and septicemia which are usually fatal.
Vaso-occlusive crises of the digits present as dactylitis, where the finger becomes severely painful, red, hot, and swollen. Abdominal vaso-occlusion usually mimics the acute abdomen in pain severity. Acute chest syndrome may present as chest pain, cough, leukocytosis, tachypnea, and respiratory difficulty in young children. These may be fatal. Sickle cell anemia patients may also present with splenic sequestration crisis. This condition occurs when sickle-shaped cells get entangled in the splenic pulp, leading to severe anemia with a rapidly enlarging spleen. The most critical central nervous system condition caused by sickle cell disease is a stroke. Retinal hemorrhages with visual loss is also a critical condition.
Repeated cycles of hemolysis lead to increased pigment load. This increase results in pigmented stone formation in the gallbladder inciting cholelithiases. Sickling in the renal vasculature causes isosthenuria, a condition where the kidneys lose their ability to concentrate the urine appropriately. It also occludes the penile vasculature which causes a prolonged, painful erection known as priapism. Avascular necrosis of the long bones, particularly the head of the femur, is also a troublesome condition. It involves the slow and gradual destruction of the articular surface of the femoral head, which requires hemiarthroplasty to restore mobility.
Aplastic crises are another significant manifestation of sickle cell disease. Here, when the presence of parvovirus B19 challenges an already stressed bone marrow, it fails to generate the appropriate number of RBCs which results in severe anemia.
Evaluation
Newborns with a family positive for sickle cell disease should undergo a screening test. This screening test for sickle cell hemoglobin is mandatory in the United States. Prenatal diagnosis in genetically prone fetuses can be made by using the chorionic villus sampling technique or by amniocentesis.[7]
A complete blood count with a peripheral picture is the initial test. Here, a reduced RBC number, reduced reticulocyte count, variable mean corpuscular volume, increased leukocyte count, reduced ESR, and the presence of the sickle-shaped cells in the periphery usually indicate the diagnosis of sickle cell disease. The presence of Howell-Jolly bodies indicates functional asplenia in the patient. Subsequent hemoglobin electrophoresis confirms the diagnosis of sickle cell disease if the concentration of sickle cell hemoglobin is more than 90% of the total hemoglobin and the fetal hemoglobin comprises the rest of the hemoglobin isotype. However, if the concentration of the sickle cell hemoglobin is at or around 45%, it indicates the presence of the sickle cell trait rather than the disease itself.
A urine analysis should be performed to rule out a urinary tract infection as a cause of hematuria and identify isosthenuria in sickle cell disease patients.
If a patient presents to the emergency department with an acute vaso-occlusive episode and the diagnosis of sickle cell disease is not yet established, one can administer an instant sickling test which may test positive for sickle cell hemoglobin. However, this test has a limiting factor. It can not differentiate between heterozygous and homozygous states of sickle cell hemoglobin.
Arterial blood gases are required to monitor the pulmonary functions in case of acute chest syndrome. Serial arterial blood gases shall be obtained to monitor the severity of the pulmonary crises.
A chest X-ray shall be performed in patients with respiratory signs and symptoms, but it may be normal in the early phase of acute chest syndrome. Plain radiography of the peripheries is used in identifying acute and subacute marrow infarctions, as well as observing old infarcts. In the case of osteomyelitis, early radiographs are not useful because they do not show significant changes. However, in the subsequent two weeks, the plain radiographs may reveal the destruction of the bone, periosteal bone formation, sinus tract, and sequestra.
MRI scans are of paramount importance in diagnosing osteomyelitis and avascular necrosis of the femoral and humeral heads. Technicium-99 scans are also used to detect osteonecrosis.
Treatment / Management
The treatment of sickle cell disease has seven major goals:
- Management of vaso-occlusive crises
- Management of chronic anemia
- Management of chronic pain
- Prevention of infections
- Prevention of complications
- Prevention of stroke
- Detection and treatment of pulmonary hypertension
Pharmacotherapy of sickle cell disease usually revolves around preventing its complications. Hydroxyurea is an antimetabolite known to increase the fetal hemoglobin levels in the circulation of RBCs; fetal hemoglobin keeps hemoglobin in soluble form and prevents polymerization of the sickle cell hemoglobin, thus preventing most complications of sickle cell disease.[8],[9],[10]
Opioids, NSAIDs, steroids, antiemetics, tricyclic antidepressants, and antibiotics are all used to cure or ameliorate the complications of the disease.
Vaccines, particularly against encapsulated bacteria, are helpful in preventing life-threatening infections as sickle cell disease patients are usually functionally asplenic. Folic acid supplementation prevents macrocytic anemia.
A bone marrow transplant is curative in sickle cell disease patients.
A blood transfusion, particularly in aplastic crises, is also essential.
Differential Diagnosis
Sickle cell disease is typically suggested by the presence of chronic hemolytic anemia along with sickle cells in the peripheral smear. Electrophoresis confirms the diagnosis. It can also elucidate other hemoglobinopathies like beta thalassemia, Hemoglobin C disease, or Hemoglobin SC disease. Hemoglobin C disease is a condition where lysin replaces position 6 glutamic acid. Hemoglobin SC is a combined variant of hemoglobin C and sickle cell disease, where 50% of the patient's hemoglobin is sickle cell hemoglobin, and rest is hemoglobin C. Hemoglobin SC is a disorder which has a low level of sickling when compared to typical sickle cell disease.
Prognosis
There is no cure for sickle cell disease and the prognosis in most patients is guarded. Most patients require multiple admissions for sickle cell crisis and the goal is to ensure a good quality of life.
Vaso-occlusive crises lead to chronic pain, disability, economic loss, and depression. When the disease strikes weight-bearing bones, the patients are not able to ambulate. Premature deaths are common and most patients remain disabled for life.
Prognostic factors for poor outcomes include:
- Hemoglobin less than 7 g/dl
- Hand-foot syndrome
- Elevated WBC in the absence of an infection
Complications
- Chronic pain
- Acute chest syndrome
- Priapism
- Aplastic crises
- Splenic sequestration
- Osteomyelitis
- Delayed growth and development
- Hand and foot syndrome
- Brain hemorrhage
- Heart disease
- Cholelithiasis
- Renal failure
- Proliferative retinitis
- Leg ulcers
- Avascular necrosis
- Pulmonary hypertension
Postoperative and Rehabilitation Care
Although the patient can exercise, most have no exercise endurance. The bone pain may also limit activity. During walking or exercise, the patient should be encouraged to drink fluids.
Deterrence and Patient Education
The key to improved outcomes is patient education. The earlier one seeks medical help, the better the outcomes and thus patients should seek help in the presence of:
- Fever
- Abdominal or chest pain
- Persistent headache
At the same time, patients should avoid:
- Tobacco
- Alcohol
- Illicit drugs like cocaine
- Seeking care from multiple healthcare institutions
Patients should be urged to:
- keep hydrated
- Eat a healthy diet
- get immunized
- Take hydroxyurea as prescribed
- Follow up with a clinician
Enhancing Healthcare Team Outcomes
Sickle cell anemia is a severe genetic disorder with very high morbidity and mortality. The disease usually manifests early in life and can present with several types of occlusive crises. Because the disorder affects almost every organ system it is best managed by an interprofessional team that is dedicated to sickle cell disease. The key is preventing sickle cell crisis and repeated hospitalizations. The sickle cell nurse should follow all outpatients and make appropriate recommendations depending on symptoms.
Screening for sickle cell anemia is mandatory at birth in the United States, allowing for early diagnosis and treatment. However, as the population ages, chronic complications like pulmonary hypertension are emerging with very high morbidity and mortality. The consensus by experts is that sickle cell should be managed by an interprofessional group of healthcare professionals, including physical therapists, psychiatrists, social workers, nurses, pharmacists, substance abuse counselors, pain counselors, and rehabilitation specialists.[11],[12] [Level V] Anytime there is a fever, an infectious disease consult should be made promptly.
An orthopedic surgeon should be consulted for hip pain or difficulty with gait. A radiologist is essential for obtaining samples from bone if osteomyelitis is suspected. These patients need thorough eye exams by an ophthalmologist, and a urologist is needed to manage priapism.
The social worker should be involved early on to ensure that patients have the support they need and do not lack finances to buy medications. In addition, because many patients are depressed or have anxiety, a mental health nurse should counsel these patients.
Most of these patients are on many medications. The pharmacist plays a critical role in ensuring the patient remains compliant with medications and free from adverse drug effects. The pharmacist should report potential drug-drug interactions to the clinician managing the case.
The nurse should educate the patient on the importance of remaining hydrated, getting the right vaccinations, and ensuring follow up with the respective healthcare providers. Typically, a specialty trained oncology and hematology nurse will assist the provider in arranging close followup and compliance.
Outcomes
The outcome for most sickle cell patients is mixed. The goal is to achieve an average lifespan with minimal morbidity and mortality. However, many of these individuals die prematurely despite improvements in treatment. The morbidity is very high, and nearly all individuals experience a vaso-occlusive crisis at some point in their lives. These patients often are not able to work due to disability and live a poor quality of life with chronic pain. The leading causes of death are acute chest syndrome, pulmonary embolism, and infection. Outside of North America, the life expectancy of sickle cell patients is in the 30s and 40s. Many guidelines have been developed to manage sickle cell disease. They include penicillin prophylaxis for children, blood transfusions, and pneumococcal vaccination. The drug hydroxyurea has made it possible for patients to live longer than ever.[13][Level V]
(Click Image to Enlarge)
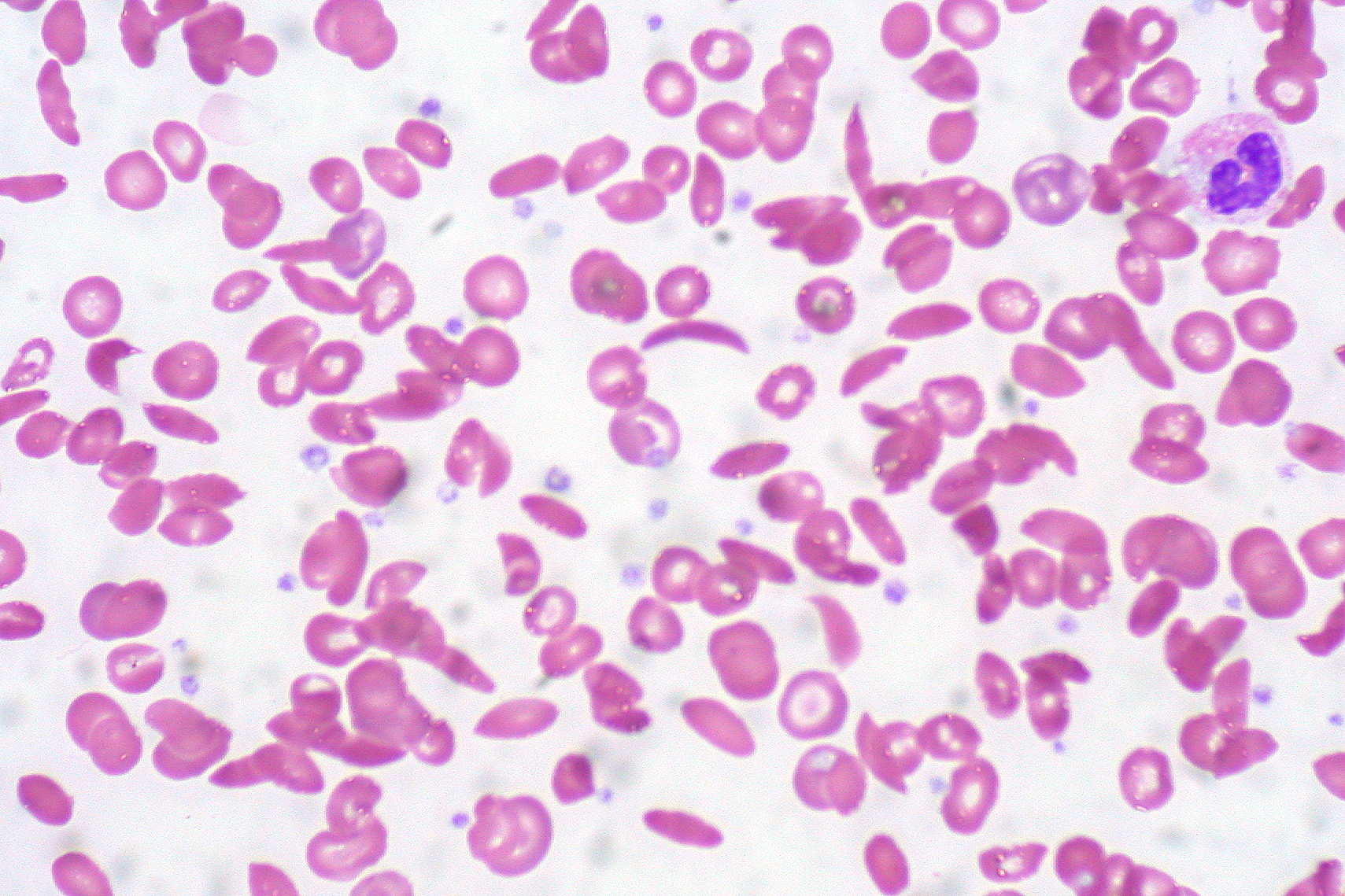
Sickle Cell Anemia, Hemoglobin C
Contributed by Ed Uthman (CC BY 2.0 https://creativecommons.org/licenses/by/2.0)