Tetralogy of Fallot
- Article Author:
- Josue Diaz-Frias
- Article Editor:
- Melissa Guillaume
- Updated:
- 8/10/2020 9:36:26 PM
- For CME on this topic:
- Tetralogy of Fallot CME
- PubMed Link:
- Tetralogy of Fallot
Introduction
Tetralogy of Fallot (TOF), historically and appropriately referred as Steno-Fallot tetralogy, was first described by the Danish physician/anatomist Dane Niels Stensen, also referenced as Nicoulas Steno in Latin, a pioneer in anatomy and geology. His work made significant contributions to the field of cardiac anatomy and pathology. The discovery of the constellation of findings that hallmark the tetralogy was first described in a short paper titled “Dissection of a Monstrous Foetus in Paris” in 1671, highlighting the unusual form in which the arteries emerge, the narrowing of the pulmonary artery, the absence of the ductus arteriosus, a subaortic interventricular septal defect, an overriding aortic canal common to both ventricles, and the physiology of fetal cardiac circulation describing how blood was redirected directly into the aorta instead of the pulmonary artery. In 1777, Eduard Sandifort, a Dutch physician, reported a case of tetralogy in a 16-month-old patient called “the blue boy.” At first, he was thought to have asthma; however, an autopsy revealed a congenitally malformed heart with no signs of ductus arteriosus or ligamentum arteriosum. Later in 1782, William Hunter, a Scottish physician, presented to the Society of Physicians in London a case of a 13-year-old boy with tetralogy and blue spells, who was discovered posthumously in 1774, along with three other cases of congenital heart disease.
Many other cases have been presented by Pulteney (1785), Abernethy (1793), Bell (1797), Dorsey (1812), and Farre (1814). The first case was reported in America at the University of Pennsylvania by Thaxter in 1816, with subsequent cases reported by Peacock (1858 and 1869), Widman (1881), and finally Fallot (1888). Etienne-Louis Arthur Fallot described in an elegant style and detail four cardinal features that differentiate it from other cyanotic cardiac conditions, emphasizing that this was not a product of chance and that cyanosis was not caused by a patent foramen ovale, as proposed by many others. He attributed it to an intrauterine pathologic process and understood that this tetralogy was essentially just one anomaly involving the pulmonary artery and the subpulmonary infundibulum, causing pulmonary stenosis, an interventricular communication, the biventricular origin of the aorta, and right ventricular hypertrophy, disqualifying the patency of the foramen ovale as a fifth anatomical association. The names used by Fallot were “La maladie bleue” (the blue disease) or “cyanose cardiaque” (cardiac cyanosis). In 1924, Maude Elizabeth Seymour Abbott, a pioneer in pediatric cardiology of Montreal, Canada, entitled it “tetralogy of Fallot.”
Etiology
The development of tetralogy of Fallot is multifactorial; it has been associated with untreated maternal diabetes, maternal intake of retinoic acid, phenylketonuria, chromosomal anomalies (trisomies 21, 18, 13), microdeletions of chromosome 22q11.2 and Alagille syndrome with JAG1/NOTCH2 mutations. Other genetic abnormalities with a predisposition for tetralogy of Fallot include mutations in transcription factor NKX2.5, methylenetetrahydrofolate reductase polymorphism, and mutations in TBX1 and ZFPM2.[1][2][3]
Epidemiology
Tetralogy of Fallot is the most common cyanotic heart condition in children who have survived untreated beyond the neonatal age, with the need for an intervention in the first year of life. It accounts for 7% to 10% of congenital defects, affecting males and females equally and occurring in 3 to 5 of every 10,000 live births.
Pathophysiology
The development of the human heart starts around the 20th day of gestation, with the fusion of the outer endocardial tubes into a single tubular structure, the cardiac tube. Subsequently, the cardiac tube folds and loops, with the development of an atrium that is cranial and dorsal, and a primitive ventricle is moving downward, ventrally and to the right. The right ventricle is the dominant chamber in the embryo and fetus, receiving 65% of the venous return, and is the main contributor to the lower part of the body, the placenta, and the lungs. The right ventricle can be described by three components: the inlet, which consists of the tricuspid valve chordae tendineae and papillary muscles; the trabeculated apical myocardium; and the infundibulum or conus. The exact embryologic process that contributes to the development of tetralogy of Fallot still is unknown, but an association that had been observed is an anterior and cephalad deviation of the infundibular septum that results in a misaligned ventricular septal defect, with an overriding aortic root causing a subsequent right ventricular outflow obstruction.
The ventricular septal defects seen in patients with tetralogy of Fallot are usually perimembranous that can extend into the muscular septum. Different factors can contribute with the right ventricular outflow obstruction, including the pulmonary valve that is usually bicuspid and stenotic, the hypoplastic pulmonary valve annulus, the deviation of the infundibular septum that causes a subvalvular obstruction, and the hypertrophy of the muscular bands in this region. The degree of the overriding aorta usually varies and receives blood flow from both ventricles. The physiological process surrounding the hypercyanotic episodes or “Tet spells” in tetralogy of Fallot consist of either a decrease in systemic vascular resistance or an increase in pulmonary resistance contributing to a right-to-left shunt across the ventricular septal defect, causing marked desaturation.[4][5][6]
History and Physical
Clinical presentation varies based on the severity of the right ventricular outflow tract obstruction, more commonly presenting as neonates with a certain degree of cyanosis. In some patients cyanosis present months later in life, when the rate of obstructions worsens. On auscultation, patients have a normal first heart sound with a single second heart sound that is loud. The greater the degree of obstruction, the more prominent the murmur, usually described as crescendo-decrescendo with a harsh systolic ejection quality and best heard at the left mid to upper sternal border with posterior radiation. Sometimes the murmur can have a regurgitant quality, and an early systolic click may be auscultated along the left sternal border. A prominent ventricular impulse and a systolic thrill may be appreciable, hepatomegaly is uncommon, and if there are prominent pulses present, it may be an indirect indication of a patent ductus arteriosus or aorticopulmonary collaterals. Other common associated cardiac abnormalities found are right aortic arch (25%), abnormal coronary arteries, significant aorticopulmonary collaterals, patent ductus arteriosus, multiple septal defects, and occasionally, aortic valve regurgitation.
“Tet spells” or hypercyanotic episodes present during the infancy or toddler age and decrease after 4 to 5 years of age. Dehydration or agitation commonly precipitate tet spells, and if patients do not receive prompt and adequate treatment, they can develop severe cyanosis and hypoxia that, subsequently, can cause syncope and even death. Rarely, patients present with clubbing; this is usually seen in those with severe, longstanding cyanosis.
Evaluation
Up to 50% of patients are diagnosed antenatally by fetal echocardiography, anticipating the need for postnatal prostaglandin therapy if there is evidence of severe right ventricular outflow obstruction. Useful studies to help with diagnosis and evaluation include chest radiograph, electrocardiogram, and echocardiogram. Chest radiographs usually show a normal-size heart silhouette, with an upturned apex and a concave main pulmonary artery segment, commonly known as “boot-shaped.” On the electrocardiogram, it is common to see signs of right atrial enlargement and right ventricular hypertrophy showing right axis deviation, prominent R waves anteriorly and S waves posteriorly, upright T wave in V1 (abnormal after 7 days of life up to 10 years of age) and a qR pattern in the right precordial leads. Among imaging studies, an echocardiogram is the gold standard, addressing the anatomy and severity of the right ventricular outflow obstruction, the location and number of ventricular septal defects, and assessing associated anomalies or variants with the coronaries arteries and the aortic arch. Cardiac magnetic resonance imaging can be used and is particularly useful in adults with repaired tetralogy of Fallot. Cardiac catheterization is not commonly used but can help to assess the level of obstruction, pulmonary stenosis or hypoplasia, coronary artery anatomy, and the presence of collaterals and accessory septal defects.
The most frequent causes of mortality in patients with no surgical intervention include hypoxic spells (68%), cerebrovascular accidents (17%), and brain abscesses (13%). Within the first year of life, 25% of infants with severe right ventricular outflow tract obstruction die if left untreated, 40% by three years of age, 70% by 10, and 95% by 40.[7][8]
Treatment / Management
Neonates with severe right ventricle outflow obstruction presenting with profound hypoxemia and cyanosis may need prostaglandin therapy to maintain ductal patency and pulmonary flow before surgical repair. Tet spells require a rapid and aggressive approach including positioning (knee-chest) to increase systemic vascular resistance, oxygen therapy to cause pulmonary vasodilation and a systemic vasoconstriction, intravenous fluid bolus to improve the right ventricle filling and pulmonary flow; morphine, intravenous beta blockers to help improve the right ventricle outflow obstruction by relaxing the muscle, and intravenous phenylephrine to increase systemic afterload. If heart failure is developed, digoxin and loop diuretics are a good pharmacological therapeutic option.[9][10][11]
Following the recommendation of the American Heart Association guidelines, all patients with unrepair cyanotic congenital heart disease should receive subacute bacterial endocarditis prophylaxis for dental procedures, respiratory procedures, or infected skin procedures. Other reasons for prophylaxis are prosthetic cardiac valves, previous history of endocarditis, and completely repaired congenital heart disease with prosthetic material or device for 6 months post-procedure.
The comprehension that tetralogy of Fallot involves only the subpulmonary infundibulum, “the monology of Stensen,” had important diagnostic and surgical repercussions. With the recognition of tetralogy of Fallot, the age of cardiac surgery to repair congenital heart defects took place. The first surgical treatment was achieved at John Hopkins with Dr. Helen Taussig. She observed that children with cyanotic heart conditions had a better prognosis if the ductus arteriosus remained open. In 1939, after reading the first report of a successful closure of a patent ductus arteriosus by Dr. Robert Gross, she thought of creating a ductus for cyanotic children. She approached Dr. Alfred Blalock and Vivien Thomas, Blalock’s laboratory surgical technician, and proposed a surgical solution for tetralogy of Fallot. In 1944, Dr. Blalock, with Thomas by his side, successfully achieved an artificial ductus through a left anterolateral thoracotomy, anastomosing the proximal end of the left subclavian artery to the left pulmonary artery in a critically ill 15-month-old female with cyanosis and a weight of 4 kg. The Blalock-Taussig shunt or systemic to pulmonary arterial shunt, originated by Blalock, Taussig, and Thomas marked the genesis of the era of congenital heart surgery, becoming an acceptable palliative therapeutic option for cyanotic defects with decreased pulmonary blood flow. Most congenital surgeons perform a modified BT shunt via posterolateral thoracotomy or median sternotomy or a central shunt via a median sternotomy. The modified BT shunt uses a prosthetic tube graft (made of polytetrafluoroethylene) that is interposed between a systemic artery and the pulmonary artery, which differs from what was first described as a direct anastomosis more likely to thrombose.
From 1950 to 1970 there was an increased understanding of tetralogy of Fallot, with standardization and advancement of surgical repairing techniques conducting a cardiopulmonary bypass, and a better postoperative management translating into a better survival rate (85% to 90%) and a decreased in perioperative mortality by the early 1960s, from 60% to 7% to 14%. Until the 1970s, very few surgeons were performing the complete repair in children younger than 3 to 5 years of age. Bonchek and Starr concluded that it was beneficial to perform a complete repair earlier in life, preventing a worse obstruction from fibrosis and undergrowth of the right ventricle outflow tract. Barrat-Boyes, Kirklin, and Castaneda contributed with the support of an early complete repair in neonates and infants with low mortality. At present, early survival after a complete primary repair in large centers is reported to be between 98% to 100%. Despite this fact, primary complete repair in neonates and infants, in general, is still controversial. Since the 1970s, surgeons have recommended the complete repair by 6 months of age and no later than 12 months, for nonductal dependent, asymptomatic infants. Controversy lays within symptomatic neonates undergoing a primary repair versus a two-stage procedure starting with a shunt placement. Relative indications for a shunt include those patients with severely hypoplastic pulmonary arteries, anomalous left anterior descending from a right coronary artery crossing the right ventricle outflow tract, or associated non-cardiac anomalies. Those who support complete repair think it promotes normal somatic growth and development, elimination of chronic hypoxemia, improved late ventricular function, decreased incidence of late dysrhythmias, and lower risk of hypercyanotic spells and their sequelae. Those opposed believe there is a higher incidence of the transannular patch and long-term consequences (pulmonary insufficiency), suggesting that a two-stage repair may increase the growth of the pulmonary valve and branch pulmonary arteries, decreasing the chance of a transannular patch. Fraser et al., after trying to make an individualize surgical strategy, found no significant difference between primary and two-stage repair regarding time to extubation, intensive care unit stay, or hospital length stay.
Transannular patching and ventriculotomy started as another therapeutic option for tetralogy of Fallot associated with pulmonary insufficiency and right ventricle dysfunction presenting with exercise intolerance, ventricular dysrhythmias, and a small incidence of sudden death. In 1963, the first transatrial transpulmonary repair by Hudspeth and colleagues was performed. This approach was revisited in the 1990s and is frequently used today in all age groups with a survival rate of greater than 99% and a low incidence of early intervention. In 1978, a monocusp valve was first described by Zavanella et al. as an innovation to create right ventricular outflow tract competence and decrease or prevent pulmonary insufficiency in a patient needing a transannular patch. Monocusp valves made from autologous or bovine pericardium, allograft pulmonary valve cusp, or polytetrafluoroethylene membrane became popular in the 1990s because they reduce pulmonary insufficiency with long-term survival of 98%, no progression greater than a moderate insufficiency in 53%, and no reoperation within ten years in 88%. Valved right ventricle to pulmonary artery conduits are used in patients with tetralogy of Fallot and pulmonary atresia or those who require reoperation for severe pulmonary insufficiency or recurrent stenosis. Pulmonary or aortic homografts are used in infants with the risk of reoperation of 50% in 5 years and when used with neonates in 3 years. Heterografts valved (bovine jugular vein graft) and autologous pericardial valved conduits are other alternatives.
Differential Diagnosis
During “Tet spells” or if the patients develop heart failure, they can present with signs and symptoms of respiratory distress, cyanosis, failure to thrive, making another diagnosis a plausible cause, such as bronchiolitis, pneumonia (viral/bacterial), pneumothorax, or severe pulmonic or aortic stenosis. Other cardiac malformations that can cause similar symptoms are those causing right-to-left shunt, including complete (d-) transposition of the great arteries with pulmonary stenosis; double outlet right ventricle, including Taussig-Bing anomaly; tricuspid atresia; Ebstein anomaly; and pulmonary atresia with intact ventricular septum. Causes of the bi-directional shunt that can also have similar symptoms are total anomalous pulmonary venous return, hypoplastic left heart syndrome, and single ventricle and truncus arteriosus.
Complications
Short Term Complications
Common complications in the immediate postoperative period are residual ventricular septal defects, as well as persistence of right ventricular outflow obstruction. Arrhythmias can follow tetralogy repair, with risk of ventricular tachycardia, atrial fibrillation/flutter, and intra-atrial reentrant tachycardia. ECG will usually appear with a right bundle branch block or left bundle branch block pattern associated with wide complex tachycardia. Sudden cardiac death can present with post-repaired patients. Risk factors for tachyarrhythmias and sudden cardiac death include older age at repair, male gender, transient complete heart block beyond postoperative day three, and QRS duration greater than 180 miliseconds.
Long-Term Complications
Adult patients with congenital heart disease are increasing in an approximate estimate of 5% per year, surpassing the pediatric population. Longterm consequences seen with these patients include right ventricular volume overload from pulmonary insufficiency, right ventricular aneurysm from outflow patch or from ventriculotomy, distal pulmonary artery obstruction, ventricular hypertrophy, chamber enlargement, biventricular dysfunction, and aortic root dilation and insufficiency. The three leading causes of mortality in patients with repaired tetralogy of Fallot are arrhythmia, heart failure, and complications from reoperations. Risk of sudden death increases after 30 years of procedure to 6% to 9%; some of the factors associated with this risk are QRS duration greater than 180 miliseconds, older age at repair (greater than 3 years), significant pulmonary valve or tricuspid valve regurgitation, history of syncope, multifocal premature ventricular contractions, and ventricular tachycardia. The most common indication for reoperation is pulmonary insufficiency, and criteria for pulmonary valve replacement have been based on the severity measured by the regurgitant fraction on magnetic resonance or CT scan. Parameters seen with these studies are right and left ventricular end-systolic and end-diastolic volume indices, ejection fractions, and presence of aneurysm causing obstructive outflow. Patient can have exercise intolerance, signs and symptoms of heart failure, syncope, and sustained ventricular tachycardia. Pulmonary valve replacement also can be obtained by transcatheter pulmonary valve approach.
Pregnancy Complications
Women who had complete repair of tetralogy of Fallot have similar outcomes in comparison with the general obstetric population. Increased pregnancy complication are related to the level of pulmonary hypertension and the severity of the pulmonary regurgitation with right or left ventricular dysfunction. Females with moderate right ventricle hypertension or those who have had a palliative shunt have an increased risk for fetal demise. Offspring of women with tetralogy carry a risk of congenital heart disease in 3% to 5% in comparison with 0.8% of the general population. If 22q11 deletion is present, the chance of transmitting the affected chromosome is 50%, with high risk of having an associated congenital heart defect.
Enhancing Healthcare Team Outcomes
The diagnosis and management of TOF is with an interprofessional team that includes a pediatrician, pediatric cardiologist, cardiac surgeon, and radiologist. In general, all children with TOF need surgery; the timing may vary depending on symptoms. In the US, the majority of infants born with TOF undergo a primary repair within the first 12 months of life. The outcomes for TOF patients are good but after two decades a significant number of them will require pulmonary valve replacement. Unlike the past, today there is a percutaneous method of implanting a pulmonary valve, but long term results are not known. After surgery, most children remain free of symptoms. It is important to remember that surgery for TOF is not curative but palliative; the structural disease continues to progress at a variable rate. [12][13][14](Level V)
(Click Image to Enlarge)
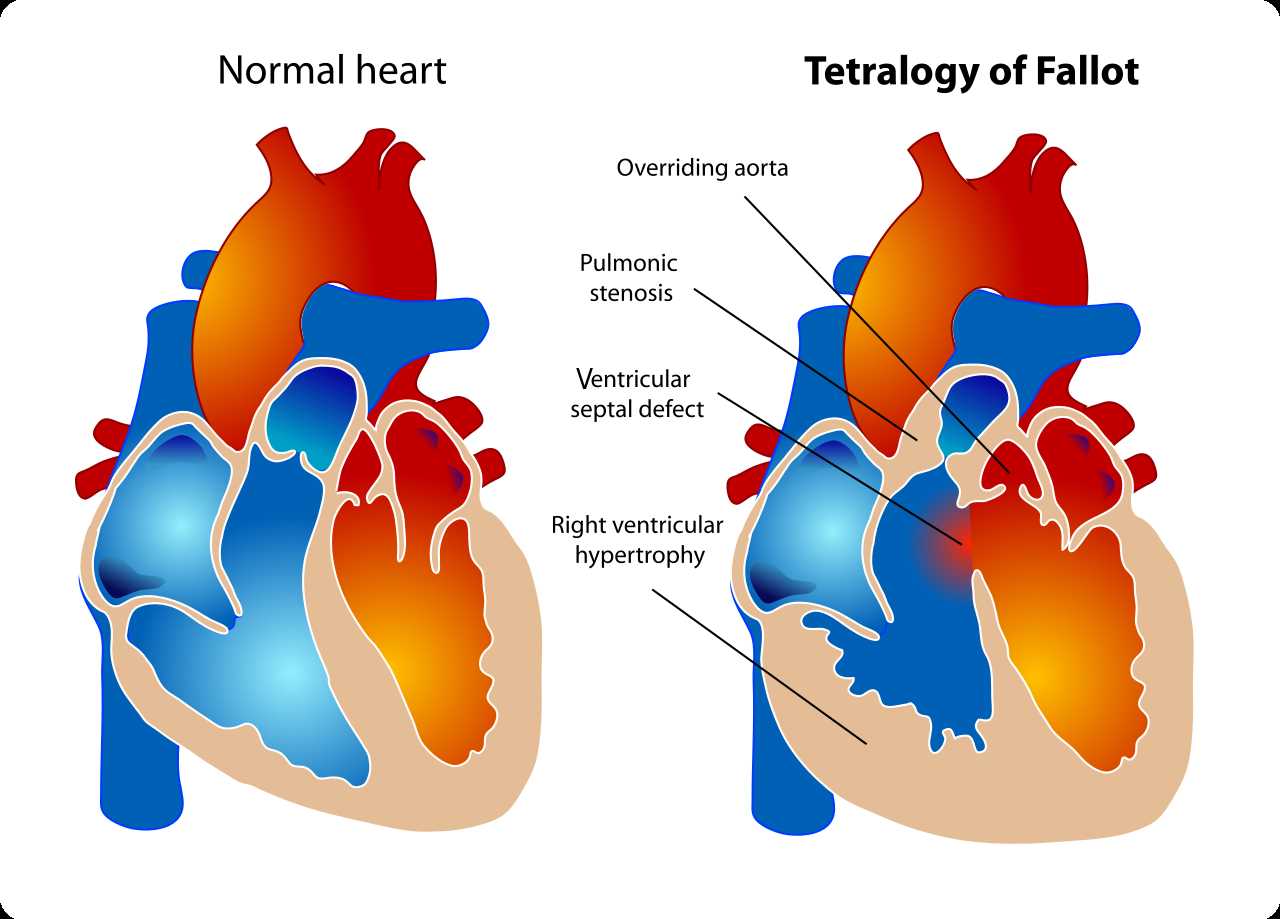
A diagram showing a healthy heart and one suffering from the tetralogy of Fallot, which constitutes four different malformations.
Contributed by Wikimedia Commons, LadyofHats (Public Domain)
(Click Image to Enlarge)

tetralogy of fallot
Contributed by S Bhimji MD