Physiology, Blood Brain Barrier
- Article Author:
- Rohin Gawdi
- Article Editor:
- Prabhu Emmady
- Updated:
- 9/27/2020 9:16:39 AM
- For CME on this topic:
- Physiology, Blood Brain Barrier CME
- PubMed Link:
- Physiology, Blood Brain Barrier
Introduction
More than 100 years ago, Ehlrich (1885) first described the existence of the blood-brain barrier (BBB). The blood-brain barrier is an entity located between the blood and brain interstitium, which composed of cerebral endothelial cells (CECs), basal lamina, astrocytic end-foot processes, and pericytes. Tight junctions connect endothelial cells and surrounded by pericytes, astrocytes, and basal lamina.[1] Astrocytes project end-foot processes along with the perivascular space, and pericytes cover the basal lamina of the endothelium and contribute to the structural integrity of the BBB.[2] Astrocyte-endothelial co-culture studies showed that several receptors, transporters, and ligands are involved in the bidirectional induction involved in BBB maintenance.[3] Absence of fenestration, the presence of intercellular tight-junctions (TJs), low level of non-specific transcytosis (pinocytosis) and paracellular diffusion of hydrophilic compounds, expression of membrane receptors and transporters which are responsible for the active transport make brain ECs significantly different from other ECs reside outside the brain. Tight junctions are composed of trans-membranous proteins, including occludin, Claudin-3, Claudin-5, and the cytoplasmic proteins Zona Occludens 1, 2, and 3 (ZO-1, ZO-2, ZO-3).[4][5][6] These tight junctions are particularly crucial for preventing the paracellular diffusion of hydrophilic solutes. The basal lamina of the cerebral endothelium is produced by ECs and astrocytes and composed of collagen IV, laminin-1,2,4, and 5, glycoproteins, and proteoglycans. Several basal lamina proteins, matrix metalloproteinases (MMPs) and their inhibitors, the tissue inhibitor of metalloproteases (TIMPs) have a role in the dynamic regulation of the BBB in physiological as well as inflammatory conditions.[4] Substances may cross the BBB primarily by two mechanisms, in between the endothelial cells or across the luminal membrane of the endothelial cell, cytoplasm, and then passing across the abluminal side of the endothelial cell into the brain interstitium. The former is called paracellular transport, and the later is transcellular transport. Tight junctions between endothelial cells typically regulate paracellular transportation. Transcellular transport occurs through passive and active mechanisms. Passive transport affected by physicochemical properties such as molecular weight, electrical charge, and lipophilicity and is typically limited to small, lipophilic molecules that are less than 500 Daltons in size. Nutrients and proteins which are larger and less lipophilic transported by active transport mechanisms. Glucose is transported by carrier-mediated transport (CMT) via the GLUT-1 protein, insulin, which undergoes receptor-mediated transcytosis (RMT), and albumin follows adsorptive-mediated transcytosis (AMT).
Issues of Concern
The BBB tightly controls the entrance of molecules from the plasma into the CNS and plays a critical role in proper CNS function. BBB dysfunction has correlations with numerous chronic degenerative neurologic disorders such as Alzheimer disease (AD), amyotrophic lateral sclerosis (ALS), multiple sclerosis (MS), and Parkinson disease (PD), as well as acute CNS disorders like traumatic brain injury (TBI) and stroke.[7]
Cellular
Various junctional molecules exist between CECs, resulting in low rates of transcytosis. TJs, which are closest to the apical membrane, consist of various claudins and occludin proteins. These proteins play a critical role in the regulation of paracellular diffusion of ions and solutes.[8] Prior studies on the experimental autoimmune encephalomyelitis (EAE) model of MS in mice have shown that claudin-1 overexpression at the BBB produced therapeutic sealing.[9] While there was a prior hypothesis that claudin-5 was the most dominant TJ protein contributing to paracellular tightness, further in vitro studies showed that claudin-5 knockout mice had ultrastructurally normal BBBs. In vivo studies on humans have shown an abundance of claudin-11 and -25, though only claudin-11 contributed to paracellular tightness while claudin-25 contributed to the maintenance of TJ strand morphology. These results indicate the involvement of other TJ-associated marvel proteins (TAMPs) in the maintenance of paracellular tightness seen in the BBB.[10] Zona occludens (ZO) -1, -2, and -3 and act as scaffolding proteins and connect intercellular TJ proteins to actin and cytoskeletal filaments within CECs.[11] These filaments and actin are recruited to scaffolding proteins by the dystrophin complex.[12] Further from the apical side of the CECs, endothelial cell adhesion molecules (ESAM) and JAM-A, -B, and -C behave as junctional molecules that play a role in regulating transendothelial leukocyte migration and maintenance of junctional tightness.[13] Gap junctions play a role in junctional tightness and enable intercellular communication between CECs via the formation of Connexin (CX)-37, -40, and -43 hemichannels.[14]
Adherens junctions are predominantly present at the basolateral membrane of CEC junctions and perform a variety of functions. VE-cadherin and platelet endothelial cell adhesion molecule-1 (PECAM-1) both form homophilic CEC-CEC contacts roughly 20 nm wide. These are bound to CEC cytoskeletal elements by adhesion proteins (AJs), which play a role in receptor signaling and leukocyte transendothelial migration. Tyrosine phosphorylation of VE-cadherin is also crucial to leukocyte transendothelial infiltration.[8]
Pericytes and CECs share a common basement membrane within the NVU and communicate to restrict solute and ion migration. N-cadherin proteins create peg-and-socket junctions between pericyte and CEC that resist mechanical stress and maintain junction tightness.[15] CX-43 hemichannel gap junctions perform Pericyte-CEC intercellular communication. [7] Besides, perivascular astroglial networks express CX-30 and -43, allowing astrocytes to exchange small molecules and ions with CECs directly.[16]
Development
Results of Stewart and Wiley’s quail-chick transplantation study indicated that characteristics of the BBB are not inherent to cerebrovascular endothelium, instead develop during CNS embryonic development.[17]
The pattern of BBB development has undergone mapping in murine CNS studies, although the exact timing is largely viewed as species-dependent.[7] At embryonic day E10, angioblasts from the perineural vascular plexus (PNVP) are guided through neuroectoderm by vascular endothelial growth factor (VEGF) secreted from ventricular layer neuroectoderm. VEGF activation of VEGFR-2 and co-receptor Neuropilin-1 are crucial to proper downstream signaling within embryonic CECs, resulting in initial angiogenesis and vascular patterning of nascent blood vessels that vascularize the neural tube.[18][19] Neural cells secrete Wnt ligands, which binds Frizzled and LRP-5/6 receptors on endothelial cells, activating the B-catenin-dependent intracellular pathways with the assistance of TSPAN12. Wnt/B-catenin signaling is aided by cofactors Gpr124, an orphan G protein-coupled receptor, and Reck, a GPI-anchored membrane protein.[20] [7] Gpr124 has been linked to maintenance of the BBB in pathologic conditions such as ischemic stroke and glioblastoma.[21][22] Activation of B-catenin stabilizes VE-cadherin and translocates to the nucleus, resulting in the upregulation of TJ component claudin-3, death receptors DR6 and TROY, and glucose transporter GLUT-1 while downregulating genes associated with vessel fenestration such as plasmalemma vesicle-associated protein (PLVAP).[23][24][25] Platelet-derived growth factor-BB (PDGF-BB) from endothelial cells binds to PDGFR-B receptor on pericytes, triggering intracell signaling pathways that result in the production of basement membrane laminins and vitronectin. These proteins support and stabilize endothelial tube formation.[26] Other pericyte-endothelial cell interactions are critical to the growth and maintenance of the BBB, such as endothelial-derived TGF-B-activin receptor-like kinase signaling and pericyte-derived angiopoietin-Tie-2 signaling.[27] The primitive BBB is fully formed in mice by embryonic day E15, although further maturation occurs throughout gestation and briefly after birth in mice.[7] In the maturation phase, astrocytes join the newly formed neurovascular unit (NVU) and produce astrocytic endfeet and glial limitans that envelope penetrating arterioles. Astrocytes also produce laminin alpha 1 and 2, which helps the basement membrane stabilize pericytes. Astrocytes also secrete SHH, leading to maturation of endothelial TJs.[28] Sonic hedgehog (SHH) gene signaling is critical for the establishment of cell polarity in the body. SHH acts on patched homolog 1 (PTC-1) receptors on endothelium, resulting in upregulated expression of TJ elements occludin, claudin-3 and 6, VE-cadherin, and junctional adhesion molecule-A (JAM-A) and downregulation of inflammatory mediators such as IL-8, CCL-2, and ICAM-1.[29] This action results in further reduced fenestration of the existing endothelial layer.[7]
Organ Systems Involved
BBB is made up of tight junctions(TJs) between endothelial cells and is the central element of the neurovascular unit (NVU). These NVUs exist within the capillaries and post-capillary venules that vascularize the CNS, including the most parts of the brain and the spinal cord. A notable exception is a group of specialized neuroepithelial structures within the CNS: the circumventricular organs (CVOs). Unlike the remainder of the CNS, CVOs are vascularized by fenestrated capillaries, which allows bidirectional communication between blood and brain. Grouped at the midline of the brain proximal to the third and fourth ventricles, CVOs perform regulated secretory and sensory functions that the remainder of the CNS cannot play that role due to the BBB.[30] There are seven significant CVOs, grouped by function into secretory and sensory organs. The four secretory CVOs are the median eminence, neurohypophysis, pineal gland, and Subcommisural organ; the three sensory CVOs are the area postrema, subfornical organ, and the organum vasculosum of the lamina terminalis. These structures and their fenestrated capillaries play critical roles in neuroendocrine function.[31] Recent MRI studies have also indicated that while the BBB regulates blood-brain interaction, the brain utilizes lymphatic vessels in the dura mater to communicate with the peripheral immune system directly, which has been evident to play a role in CNS immune response and toxin transport.[32]
Function
The essential function of the BBB is to tightly regulate ion and solute transport between the intravascular plasma and the CNS, which occurs via molecular exchange pathways that transport molecules from blood-to-brain and brain-to-blood. These transport mechanisms have been isolated and verified via transcriptomic experiments in rodent CNS studies. Not all molecules require transport mechanisms; gases such as carbon dioxide and oxygen and lipophilic molecules under 400Da diffuse freely across the BBB. While astrocytes play a significant role in stabilization and maintenance of BBB integrity, the CECs and pericytes contain the most critical BBB transport mechanisms and pathways. While other studies have analyzed the role of transporters present on astrocyte end-foot processes, transcriptome studies are still underway.[33]
Mechanism
Passive Transport Mechanisms
Endothelial carrier-mediated transporters (CMT) are the primary mechanism for substrate-specific transport of carbohydrates, hormones, amino acids (AA), fatty acids (FAs), nucleotides, and vitamins across the BBB.[7] GLUT-1, a carbohydrate transport uniporter present on CECs, moves glucose and other hexose sugars down the concentration gradient. Although the transporter is capable of transporting hexose sugars into and out of the brain, glucose concentration is lower within the CNS than within plasma, resulting in a mostly unilateral movement of sugars. The importance of GLUT-1 transporter function to CNS function has been demonstrated by the abundance of SLC2A1 gene transcripts in brain endothelium and was further evidenced by results showing that SLC2A gene mutations have links to profound BBB and CNS dysfunction in knockout studies.[34] Pericytes express the facilitative glucose uniporter GLUT-10 and insulin-gated glucose transporter GLUT-4, allowing them to transport hexose sugars into brain interstitium similarly.[35] In periods of metabolic stress, such as fasting or exercise, monocarboxylate compounds such as lactate and ketone bodies can replace absent glucose as the brain’s primary energy source. Monocarboxylate transporter-1 (MCT-1) mediates the movement of these compounds from blood-to-brain. Pericytes express a similar transporter for monocarboxylate compounds, MCT-12.[36] AA transport across the BBB is largely under the mediation of a variety of residue-specific transporters that migrate molecules down the concentration gradient. Neutral essential AA, such as tryptophan and tyrosine, and cationic AA lysine and arginine are transported from blood-to-brain via the large neutral endothelial AA transporter 1 and 2 (LAT-1/2) and cationic AA transporter 1 and 3 (CAT-1/3), respectively. Because concentrations of AA in the CNS are lower than in plasma, these transporters mediate blood-to-brain entry. Excitatory AA glutamate and aspartate are present in higher levels within brain interstitium. Sodium-dependent transporters of excitatory AA (EAAT-1/2/3) mediate removal into blood, thus limiting neurotoxic effects. Nonessential AA alanine, serine, and cysteine are removed from CNS by ASCT-1/2, while similarly nonessential glycine is removed by GLYT-1 transporters. Additionally, glutamine is removed from CNS interstitium via sodium-coupled AA transporters 1, 2, 3, and 5 (SNAT-1/2/3/5). The removal of these acidic AA residues prevents the mediation of neurotoxic effects and is critical to CNS function.[37] Thyroid hormones triiodothyronine (T3) and thyroxine (T4) both require transport across the BBB to maintain proper brain development and prevent certain pathogenic mechanisms. T3 movement into the brain is mediated by MCT-8, while the T4 movement relies on organic anion transporting polypeptide 1c1 (OATP1C1) on CECs.[38] FAs are critical to CNS development and maintenance of neural function. FA transport proteins 1 and 4 (FATP-1/4) mediates luminal transport of FA into the brain, while MFSD2A, an orphan MFS transporter, is critical for supplying omega-3 FA into the brain; this compound plays a role in BBB development and functionality. Monogenic loss of MSFD2A is a notable aspect of pathology, as discussed below.[39] Two sets of sodium-independent transporters transport nucleotides: concentrative nucleoside transporters (CNT-2) and equilibrative nucleoside transporters-1,2 (ENT-1/2).[40] Organic anions and cations, such as carnitine, play a significant role in FA oxidation within neurons and require blood-to-brain transport to be used by the brain tissue. Anions require organic anion transporters (OAT), while cations are transported by organic cation transporter (OCT) molecules. It is clinically significant to note that N-methyl-4 phenyl-1, 2, 3, 6-tetrahydropyridine (MPTP), a molecule that induces parkinsonian motor impairment, is transported into the brain via OCT-1/2. Cation transport from brain-to-blood is executed primarily by plasma membrane monoamine transporter (PMAT). Choline transport occurs with equal bidirectionality via the choline transporter-like protein type 1 (CTL-1). Sodium-dependent multivitamin transporter (SMVT) transports critical vitamins into the CNS.[39] While individual AA residues are easily carried across the BBB by designated transporters, larger polypeptide and macromolecules can utilize a different transport mechanism called transcytosis to traverse from apical to basolateral membranes of the CEC. This process is referred to as the RMT, is much slower than carrier-mediated transport seen for nutrients, instead of requiring a sequence of steps including endocytosis of the molecule, intracellular vesicle trafficking, and exocytosis; this is a function retained among all endothelial cells; however, receptors for initial binding are highly-specific for a narrow range of molecules to be transported. Transferrin, insulin, and leptin each obtain entry to the BBB by binding to their respective transporters: transferrin receptor (TfR), insulin receptor (IR), and leptin receptor (LEP-R). V1 vasopressinergic receptors play a role in arginine vasopressin transport bidirectionally. It is notable to mention that albumin receptors are not present on CECs, rather albumin undergoes transcytosis via adsorptive-mediated processes like other positively charged macromolecules. TfR and IR transcytosis mechanisms are both potential targets for drug-delivery for CNS-drugs.[41] Similarly, lipoprotein receptors LRP-1/2 are expressed on CECs, and mediate efflux of beta-amyloid toxin in AD mediates through the BBB into the systemic blood flow.[37]
Active Transport Mechanisms
ATP-binding cassette (ABC) transporters utilize ATP on the luminal side of CEC, preventing CNS accumulation of neuroactive substances by actively shuttling across the BBB into plasma. Examples of these include ABC-B1/A2, BCRP, MRP-1/2/3/4/5. ABC-B1 plays a role in beta-amyloid toxin clearance to blood from the brain in AD. Additional studies have shown that ABC-B1 dysfunction or reduced expression frequently presents in chronic neurodegenerative disorders.[42] Endothelial ion transport is also an active process that maintains ion concentration within the CNS. Luminal Na+-K+-ATPase pumps are critical to Na+ influx into CNS and K+ efflux from CNS, maintaining high Na concentrations and low K concentration within the brain. Voltage-gated K+ channels Kv1 and Kir2 maintain the hyperpolarization of CECs and play a role in the regulation of cerebral blood flow. A luminal Na-K-Cl cotransporter (NKCC1) mediates the influx of Na, K+, and 2Cl molecules from plasma into CECs. At the basolateral side, this mechanism is complemented by a bicarbonate-Cl- exchanger and Na-H exchanger, resulting in Cl- and Na+ influx into CNS and H+ and bicarbonate efflux into the blood. This mechanism is critical for the pH regulation of CECs and the CNS. Na-Ca exchange cotransporter mediates the efflux of Calcium from the CNS, maintaining low levels of Calcium in the brain. Transient receptor potential channels (TRP) are expressed on CECs and regulate calcium influx into the brain by playing a role in vasodilatory endothelial-derived factors. These active ion transport mechanisms are present on both CECs and pericytes.[43]
Pathophysiology
Monogenic Diseases
These rare disorders stem from genetic mutations that lead to dysfunction or absence of critical endothelial and mural cell components within the BBB. Endothelial cells can experience loss-of-function mutations of the SLC2A1 gene, which is responsible for encoding the GLUT-1 hexose transporter. The loss of this essential transport mechanism presents with developmental delay, microcephaly, and early-onset seizure activity.[44] MFSD2A gene mutations that result in reduced/dysfunctional sodium-dependent endothelial transporters result in the inability to transport essential omega-3 fatty acids into the CNS, presenting as neuron loss and intellectual disability. Microcephaly in this patient population is often fatal.[7] SLC16A2 gene mutations on CECs can result in a rare disorder called Allan-Herndon-Dudley syndrome, which results in loss of MCT-8 and the inability to traffic thyroid hormone T3 across the BBB. This condition can result in profound intellectual and psychomotor disability and is also associated with aberrant serum thyroid studies, a possible critical clinical indication that this disorder is present.[45] Mutations resulting in aberrant or nonfunctional CCM-2, PDCD-10, and KRIT-1 proteins can result in developmental malformation of the cerebral microvasculature, resulting in thin and leaky capillaries. These vessels tend to be seen in cortical white matter and results in fragile vessels that are prone to bleeding. Loss of any of these three transporters can present as gait ataxia, focal neurologic deficits, and seizures. Patients also have a significantly increased risk of stroke. Gene mutations on basement membrane collagen genes COL4-A1 and COL4-A2 causes pathology in numerous body systems and cause small vessel disease within the CNS. Patients often experience lacunar strokes and small vessel hemorrhages. The role of these genes in BBB dysfunction requires further study, but mice models have shown that these mutations result in the propensity to vessel damage upon hemodynamic stress. OCLN gene mutations result in loss of occludin, a critical protein to the maintenance of TJ endothelial junctions. These can have profound effects on CNS, resulting in pseudo-TORCH one syndrome, defined by microcephaly, seizures, developmental delay, and gray matter calcifications seen on neuroimaging.[46] Homozygous JAM-3 gene mutations result in loss of JAM junctional molecule proteins, resulting in BBB failure and symptoms associated with weakened CNS microvasculature, such as hemorrhages and seizures. Uniquely, JAM-3 mutations may present with congenital cataracts. PDGF-B mutations on endothelial cells or PDGF-RB gene mutations on pericytes can cause a pattern of brain calcification and neuronal cell death called Fahr's Disease or idiopathic basal ganglia calcification (IBGC).[47]
Chronic Neurodegenerative Diseases
There are two primary forms of Alzheimer's disease (AD), autosomal dominant AD (ADAD), and sporadic AD. ADAD is associated with mutations on amyloid precursor protein (APP) gene and presenilin-1 and 2 genes. Mutations of these proteins result in BBB breakdown. Patients with ADAD present with early-onset AD, but only makes up only about 1% of all cases AD. In sporadic cases, the presence of Apolipoprotein E4 (APOE4) is a significant risk factor. APOE4 has shown to cause some degree of BBB dysfunction, diminishing clearance of beta-amyloid toxin across the BBB into the systemic blood flow.[48] Animal studies of ADAD have indicated that BBB breakdown actually precedes symptoms and pathologic findings. Post-mortem studies of the BBB in patients with both forms of AD show perivascular accumulation of thrombin, albumin, IgG, hemosiderin, and fibrin. These accumulations often localize alongside beta-amyloid plaques. AD cortex studies have also indicated that AD results in the collection of osmophilic material in mural cells, causing pericyte death. Confirmation is by a marked decrease in PDGF-RB expression on AD patient biopsy samples compared to control brains. Similarly, TJ proteins and basement membrane proteins dramatically decrease in patients with AD compared to controls. When still alive, patients with AD present with significantly reduced GLUT-1 transporter function and reduced LRP1, a receptor that clears beta-amyloid to the blood.[36] The overall loss of CNS vessel function is not counteracted by normal angiogenesis, although an attempted angiogenic response is present. Thus the brain in AD is incapable of replacing lost capillaries, although angiogenic factors are present in increased levels in serum.[48] While idiopathic Parkinson disease (PD) has no clear etiology, BBB dysfunction is a finding. The pattern of perivascular accumulation and CEC degeneration is quite similar to that of AD. Recent studies have offered evidence that alpha-synuclein can pass through the BBB bidirectionally, a process that occurs with increasing frequency during LPS-induced BBB breakdown, a pattern noted in the brains of PD patients. Unlike in AD, the angiogenic response in PD results in increased angiogenesis in some parts of the brain. However, this response does not occur at the substantia nigra pars compacta, putamen, or locus ceruleus, brain areas severely damaged in PD.[49] ALS has been linked to inheritance of numerous genes in rodent models, the most important of which is SOD-1 mutations, although 90% or more cases of ALS in humans are sporadic. Post-mortem studies of patients with ALS show dramatic loss of TJ proteins, accumulation of serum proteins, detachment of the astrocytic endfeet, and perivascular spacing indicative of BBB breakdown. Similar to PD and AD, ALS brains also have pericyte loss, most notably seen in the ventral horn of the spinal cord. BBB dysfunction and breakdown is present in several other chronic neurodegenerative disorders, such as HIV-1-associated dementia, MS, and chronic traumatic encephalopathy (CTE).[50]
Acute Neurologic Diseases
BBB dysfunction is a significant element of acute neurologic pathology, including stroke, trauma, and epilepsy. In ischemic and hemorrhagic stroke, noted BBB dysfunction had correlated with adverse clinical outcomes. BBB disruption after an ischemic stroke is a biphasic phenomenon: Phase 1 occurs approximately 30 to 60 minutes post-injury, yielding small alterations in the cytoskeleton of CECs, while Phase 2 occurs 2 to 3 days after the first, resulting in recruitment of inflammatory cells and enzymatic cleavage of TJ proteins.[51] This activity also results in increased BBB permeability to ions, solutes, cells, and proteins. Thus, damage to the BBB results in the presence of extravasated blood cells and edema in CNS parenchyma. Leukocyte infiltration into the CNS is also associated with worsened brain injury and has correlations with worse clinical outcomes. Such disruption of BBB in stroke is associated with increased risk of subsequent bleeding, which limits the utility of therapeutic options such as tPA. Similarly to ischemic stroke, damage to cerebrovascular structures in traumatic brain injury (TBI) results in activation of the coagulation cascade within vessels, leading to ischemia similar to ischemic strokes, resulting in damage to BBB, following the same biphasic pattern of infiltration and damage to BBB. In epilepsy, although the mechanism of BBB damage is unclear, TJ protein loss and IgG infiltration are seen in brain areas affected by seizures. Dysfunction of the BBB and subsequent neuronal death is strongly localized to epileptic regions, providing evidence that BBB damage plays a role in epilepsy pathogenesis.[52]
Clinical Significance
The primary clinical significance of the BBB relates to traversing the BBB for the treatment of CNS disorders. The tight regulation of molecular passage and the presence of highly efficient efflux systems results in the rejection of the vast majority of small and large molecule therapeutics from staying in the brain. Additionally, BBB damage in pathologic states can make targeting focal injury points difficult.[7] Few currently-used drugs traverse the BBB well and have enjoyed significant use in CNS pathology. One of the few, L-3,4-dihydroxyphenylalanine (L-DOPA), crosses the BBB via the neutral AA transporter LAT-1, a CMT system, and has been used to elevate CNS dopamine levels in PD.[41] Finding other drugs that successfully traverse the BBB and also have neuroactive effects has proven difficult; thus, researchers have explored different strategies to circumvent the BBB. One such approach has been physically bypassing the BBB by temporarily disrupting the CEC junctions to allow drug penetration into the CNS. Intravenous injection of hypertonic sugar solutions and focused ultrasound scans with microbubbles have each both shown the ability to grant transient rises in cerebral blood pressure, allowing drugs to traverse a disrupted BBB and access the CNS briefly.[53] These methods require much more testing, especially on their long-term safety. There are concerns that microbubbles may induce local CNS inflammatory responses that can precipitate mild ischemic injury similar to TBI.[54] Another strategy in development is the use of liposome-enclosed or polymer-coated nanoparticles that can successfully traverse the BBB. These structures would carry a negative charge that would allow undisrupted flow across the BBB or would be capable of binding BBB receptors, mediating entrance into the CNS.[55] Besides, attempts have been made to target existing CMT and RMT transport mechanisms on the BBB for drug penetration. Drugs that are transported by the CMT system tend to be small-molecule drugs, a prime example of which is L-DOPA. RMT systems are highly selective; however, they play a critical role in ensuring adequate clearance of toxins and exclusive entry of specific polypeptides. RMT systems have tremendous potential for large molecule drug delivery. A newly developed example of this mechanism is the molecular Trojan horse, which consists of drug polypeptides bound to RMT-specific antibodies. Antibody-RMT receptor binding and passage into CNS allows the polypeptide to then act on the brain. Currently, two Molecular Trojan horse drug delivery systems are undergoing clinical testing.[41]
(Click Image to Enlarge)
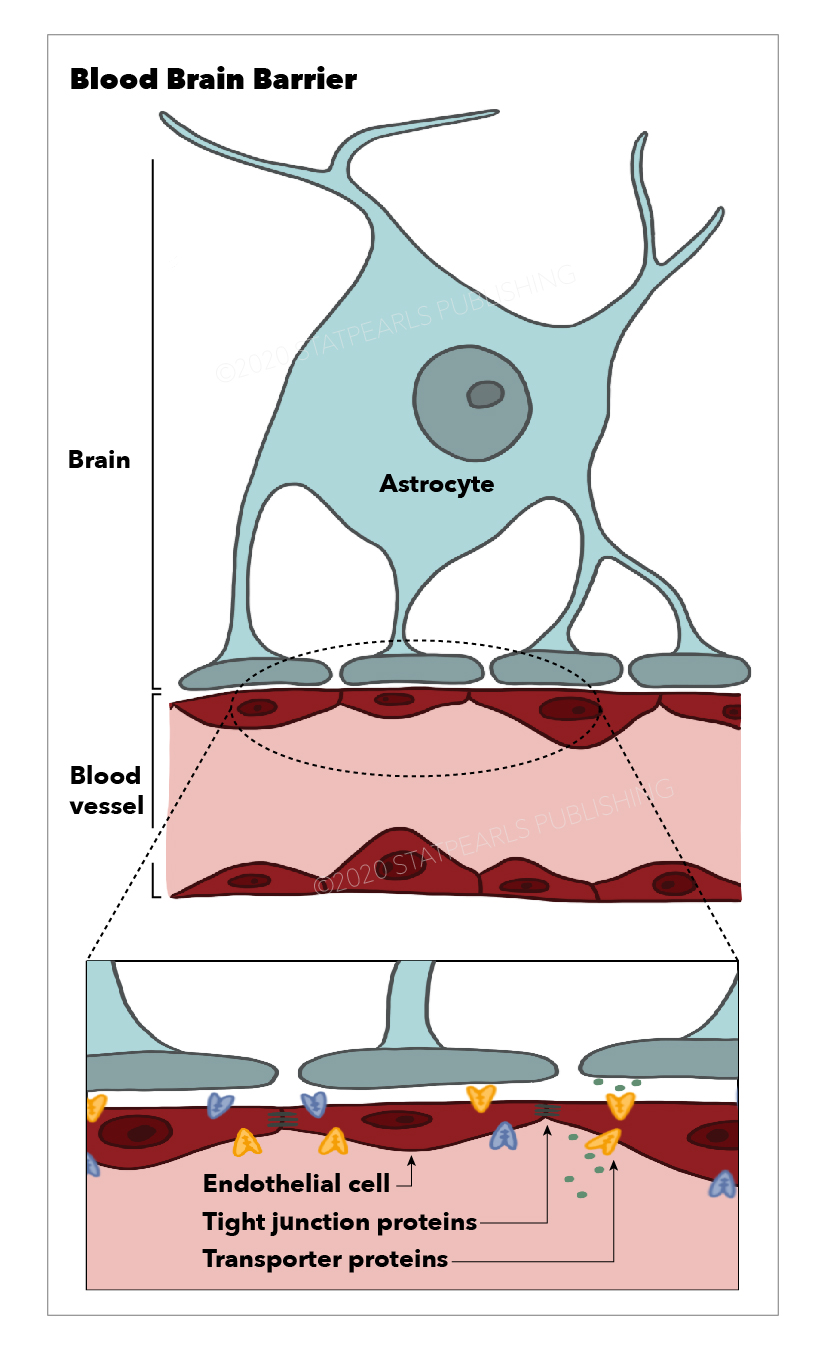
Blood Brain Barrier, Astrocyte, Endothelial cell Tight junction proteins, Transporter proteins, Blood vessel
Illustration by Emma Gregory