6-Pyruvoyltetrahydropterin synthase deficiency
| 6-Pyruvoyltetrahydropterin synthase deficiency | |
|---|---|
| Other names: Hyperphenylalaninemia due to 6-pyruvoyltetrahydropterin synthase deficiency[1] | |
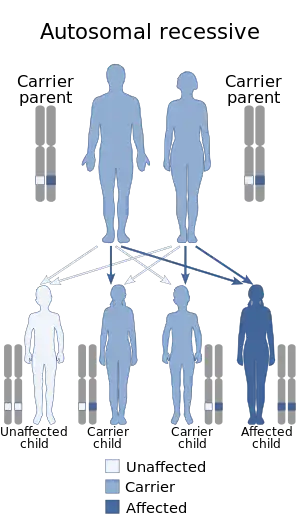 | |
| This condition is inherited in an autosomal recessive manner | |
6-Pyruvoyltetrahydropterin synthase deficiency is an autosomal recessive disorder that causes malignant hyperphenylalaninemia due to tetrahydrobiopterin deficiency.[2] It is a recessive disorder that is accompanied by hyperphenylalaninemia. Commonly reported symptoms are initial truncal hypotonia, subsequent appendicular hypertonia, bradykinesia, cogwheel rigidity, generalized dystonia, and marked diurnal fluctuation. Other reported clinical features include difficulty in swallowing, oculogyric crises, somnolence, irritability, hyperthermia, and seizures. Chorea, athetosis, hypersalivation, rash with eczema, and sudden death have also been reported. Patients with mild phenotypes may deteriorate if given folate antagonists such as methotrexate, which can interfere with a salvage pathway through which dihydrobiopterin is converted into tetrahydrobiopterin via dihydrofolate reductase. Treatment options include substitution with neurotransmitter precursors (levodopa, 5-hydroxytryptophan), monoamine oxidase inhibitors, and tetrahydrobiopterin.[3] Response to treatment is variable and the long-term and functional outcome is unknown. To provide a basis for improving the understanding of the epidemiology, genotype–phenotype correlation and outcome of these diseases, their impact on the quality of life of patients, and for evaluating diagnostic and therapeutic strategies a patient registry was established by the noncommercial International Working Group on Neurotransmitter Related Disorders (iNTD).[4]
Signs and symptoms
Movement disorders mentioned are caused by the inability to produce L-Dopa. Dopamine is a neurotransmitter which is also involved in movement. The absence of enough of this molecule causes the movement disorders in an affected individual.
Mechanism
To understand how the absence of this enzyme affects the body, we must look at the BH4 synthesis pathway. PTPS is an intermediate in this cycle and is needed to convert 7,8 - dihydroneopterin triphosphate to 6-Pyruvoyltetrahydryobiopterin. 6-Pyruvoyltetrahydryobiopterin is converted into BH4 (Tetrahydrobiopterin), but since it stops at 6-Pyruvoyltetrahydrobiopterin no BH4 is made.[5] The absence of BH4 affects the metabolism of Phenylalanine. This is the reason that PKU and PTPS deficiency share some similar symptoms. However, since BH4 is needed for much more than just the metabolism of Phenylalanine, there are other symptoms as well.
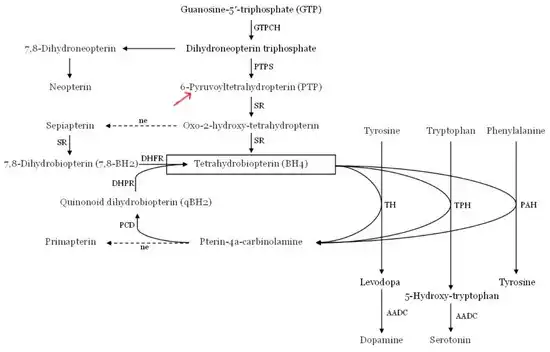 The metabolic pathway of tetrahydrobiopterin (PTPS, 6-pyruvoyltetrahydropterin synthase)
The metabolic pathway of tetrahydrobiopterin (PTPS, 6-pyruvoyltetrahydropterin synthase)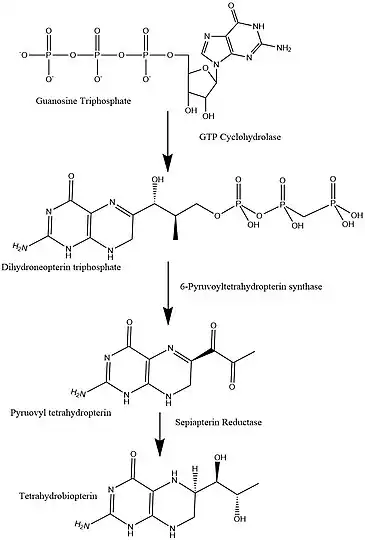 This image depicts the pathway for the synthesis of tetrahydrobiopterin, a very important cofactor in the human body.
This image depicts the pathway for the synthesis of tetrahydrobiopterin, a very important cofactor in the human body.
Structure and function of 6-Pyruvoyltetrahydropterin Synthase
PTPS is a hexamer of identical subunits. The PTPS monomer folds into a sequential four-stranded, antiparallel 𝛃-sheet. Three PTPS monomers form a 12-stranded antiparallel 𝛃-barrel by tight association between the N and C terminus of 2 adjacent subunits. The active enzyme complex is formed by two trimers in a head-to-head fashion. The active site is in between the trimer-trimer interface and has 3 monomers: A, A′, B.[6] There are three histidine residues that form the metal-binding site in the substrate binding pocket: His 23,48 and 50. Residues Cys42, Glu133, and His89 are in close proximity to the binding pocket but are not binding it. These are thought to serve as proton donors and acceptors during catalysis. The cofactor bound can be either Mg2+ or Ni2+ (Protein Database). As previously mentioned it is involved in the biosynthesis of BH4 and catalyzes the transformation of 7,8-dihydroneopterin triphosphate into 6-pyruvoyl tetrahydropterin synthase. The kinetic values for this enzyme are KM = 8.1 μM and Vmax = 120 nmol/min/mg.[7] The structure of PTPS is unique and is different than other examples of antiparallel 𝛃-barrels. It is described as being formed by "a three-fold symmetrical arrangement of subunits".[6] A similar 𝛃-barrel formation has been discovered in CKS2,[8] the difference between PTPS and CKS2 is that the formation of the latter is metal induced and reversible. The PTPS trimer is similar to a porin[6] and is hypothesized to serve as a tunnel for dihydroneopterin triphosphate.
Diagnosis
PTPS deficiency can usually be detected by the same screening test used for PKU, because both disorders result in elevated levels of Phenylalanine. This test is known as the Guthrie test and is done on babies a few days after birth. Another diagnostic method is to measure Pteridine levels in urine and to measure neurotransmitters 5-hydroxyindolacetic acid (5-HIAA) and homovanillic acid (HVA).[9]
Treatment
In terms of treatment for this condition we find that prescription of tetrahydrobiopterin is done[1]
Epidemiology
Taiwanese Chinese are more likely to get PTPS deficiency as the prevalence in Taiwanese Chinese is about 1/132,000 compared to white individuals at about 1/1,000,000. The image below illustrates frequency of different BH4 deficiency defects. This is suggested to be due to the Founder effect.[10] The smaller pie charts on the side illustrate the incidence of DHPR and PTPS deficiency around the world. As can be seen PTPS deficiency occurs most commonly in Asian countries and this makes sense because of the incidence of this disease in Taiwanese Chinese being significantly higher than any other population.
History
PTPS deficiency is not necessarily its own disease. It shares history with PKU and hyperphenylalaninemia (HPA) . Asbjørn Følling, a physician studying metabolic diseases, identified an excess of phenylpyruvate as the cause of a strange, musty odor from the urine of two Norwegian children.[11] Further research by Penrose in 1935 lead to the coining of the term, “phenylketonuria”. The foundations for dietary restrictions were laid down by George Jervis and Host Bickel which is still one of the best ways to treat PKU. However, this very practice of excluding phenylalanine from the diet was not working for other forms of HPA. This was attributed to a deficiency of tetrahydrobiopterin (BH4), a cofactor for PAH.[12] The most common form of BH4 deficiency is due to PTPS. Later on PTPS was discovered and it was learned that PKU causes HPA but not all HPA is PKU.[13]
See also
- 6-pyruvoyltetrahydropterin synthase
References
- 1 2 RESERVED, INSERM US14-- ALL RIGHTS. "Orphanet: 6 pyruvoyl tetrahydropterin synthase deficiency". www.orpha.net. Archived from the original on 1 July 2022. Retrieved 6 July 2023.
- ↑ Thöny B; Blau N (1997). "Mutations in the GTP cyclohydrolase I and 6-pyruvoyl-tetrahydropterin synthase genes". Hum. Mutat. 10 (1): 11–20. doi:10.1002/(SICI)1098-1004(1997)10:1<11::AID-HUMU2>3.0.CO;2-P. PMID 9222755. S2CID 9085242.
- ↑ Pearl PL; Taylor JL; Trzcinski S; Sokohl A (May 2007). "The pediatric neurotransmitter disorders". J Child Neurol. 22 (5): 606–616. doi:10.1177/0883073807302619. PMID 17690069. S2CID 10689202.
- ↑ "Home". Archived from the original on 2021-04-28. Retrieved 2023-05-25.
- ↑ Escalante-Acosta, Bruno Alfonso; Ramos-Brizuela, Luz Maria; Vargas-Robles, Hilda; Rubio-Guerra, Alberto Francisco (2010-09-29). "Is tetrahydrobiopterin a therapeutic option in diabetic hypertensive patients?". Integrated Blood Pressure Control. 3: 125–32. doi:10.2147/ibpc.s7479. PMC 3172060. PMID 21949628.
- 1 2 3 Nar, H.; Huber, R.; Heizmann, C.W.; Thöny, B.; Bürgisser, D. (March 1994). "Three-dimensional structure of 6-pyruvoyl tetrahydropterin synthase, an enzyme involved in tetrahydrobiopterin biosynthesis". The EMBO Journal. 13 (6): 1255–1262. doi:10.1002/j.1460-2075.1994.tb06377.x. ISSN 0261-4189. PMC 394939. PMID 8137809.
- ↑ Scherer-Oppliger, Tanja; Leimbacher, Walter; Blau, Nenad; Thöny, Beat (1999-10-29). "Serine 19 of Human 6-Pyruvoyltetrahydropterin Synthase Is Phosphorylated by cGMP Protein Kinase II". Journal of Biological Chemistry. 274 (44): 31341–31348. doi:10.1074/jbc.274.44.31341. ISSN 0021-9258. PMID 10531334.
- ↑ Parge, H.E.; Arvai, A.S.; Tainer, J.A. (1995-02-07). "Human Ckshs2 Atomic Structure: A Role for ITS Hexameric Assembly in Cell Cycle Control". Science. 262 (5132): 387–95. doi:10.2210/pdb1cks/pdb. PMID 8211159.
- ↑ "6-pyruvoyl-tetrahydropterin synthase deficiency | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Archived from the original on 2018-12-10. Retrieved 2018-12-09.
- ↑ Liu, Tze-Tze; Chiang, Szu-Hui; Wu, Sheu-Jen; Hsiao, Kwang-Jen (November 2001). "Tetrahydrobiopterin-deficient hyperphenylalaninemia in the Chinese". Clinica Chimica Acta. 313 (1–2): 157–169. doi:10.1016/s0009-8981(01)00669-6. ISSN 0009-8981. PMID 11694255.
- ↑ FollingIA. 1934. Uber Ausscheidung von Phenylbrenztraubensaure in den Harn als Stoffwechselanomalie in Verbindung mit Inbicillitat. Hoppe Seylers Z Physiol Chem 227:169.
- ↑ Blau N, Thony B, Cotton RGH, Hyland K. 2001. Disorders of tetrahydrobiopterin and related biogenic amines. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Vogelstein B, editors. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill. pp 1725–1776.
- ↑ Blau, Nenad (February 2016). "Genetics of Phenylketonuria: Then and Now". Human Mutation. 37 (6): 508–515. doi:10.1002/humu.22980. PMID 26919687.
External links
| Classification | |
|---|---|
| External resources |
|