Bafilomycin
 | |
| Names | |
|---|---|
| IUPAC name
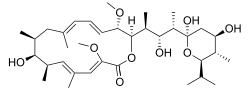
(3Z,5E,7R,8S,9S,11E,13E,15S,16R)-16- [(1S,2R,3S)-3-[(2R,4R,5S,6R)-2,4-dihydroxy-6- isopropyl-5-methyl-2-tetrahydropyranyl]-2- hydroxy-1-methylbutyl]-8-hydroxy-3,15- dimethoxy-5,7,9,11-tetramethyl-1- oxacyclohexadeca-3,5,11,13-tetraen-2-one | |
| Identifiers | |
CAS Number |
|
| ChEMBL | |
| ChemSpider | |
| DrugBank | |
| ECHA InfoCard | 100.150.187 |
PubChem CID |
|
| Properties | |
Chemical formula |
C35H58O9 |
| Molar mass | 622.83 g/mol |
| Appearance | Yellow powder |
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
| Infobox references | |
The bafilomycins are a family of macrolide antibiotics produced from a variety of Streptomycetes. Their chemical structure is defined by a 16-membered lactone ring scaffold.[2] Bafilomycins exhibit a wide range of biological activity, including anti-tumor,[3] anti-parasitic,[4][5] immunosuppressant[6] and anti-fungal[7] activity. The most used bafilomycin is bafilomycin A1, a potent inhibitor of cellular autophagy. Bafilomycins have also been found to act as ionophores, transporting potassium K+ across biological membranes and leading to mitochondrial damage and cell death.[8]
Bafilomycin A1 specifically targets the vacuolar-type H+ -ATPase (V-ATPase) enzyme, a membrane spanning proton pump that acidifies either the extracellular environment or intracellular organelles such as the lysosome. At higher micromolar concentrations, Bafilomycin A1 also acts on P-type ATPases, which have a phosphorylated transitional state.[2]
Bafilomycin A1 serves as an important tool compound in many in vitro research applications; however, its clinical use is limited by a substantial toxicity profile.[9]
Discovery & History
Bafilomycin A1, B1 and C1 were first isolated from Streptomyces griseus in 1983.[9] During a screen seeking to identify microbial secondary metabolites whose activity mimicked that of two cardiac glycosides, bafilomycin C1 was identified as an inhibitor of P-ATPase with a ki of 11 μM. Bafilomycin C1 was found to have activity against Caenorhabditis elegans, ticks, and tapeworms, in addition to stimulating the release of γ-aminobutyruc acid (GABA) from rat synaptosomes. Independently, bafilomycin A1 and other derivatives were isolated from S. griseus and shown to have antibiotic activity against some yeast, Gram-positive bacteria and fungi.[10] Bafilomycin A1 was also shown to have an anti-proliferative effect on concanavalin-A-stimulated T cells. However, its high toxicity has prevented use in clinical trials.[2]
Two years later, bafilomycins D and E were also isolated from S. griseus. In 2010, 9-hydroxy-bafilomycin D, 29-hydroxy-bafilomycin D and a number of other bafilomycins were identified from the endophytic microorganism Streptomyces sp. YIM56209.[11] From 2004-2011, bafilomycins F-K were isolated from other Streptomyces sp.[9]
As one of the first identified and most commonly used, bafilomycin A1 is of particular importance, especially as its structure serves as the core of all other bafilomycins. With its large structure, bafilomycin has multiple chiral centers and functional groups, which makes modifying its structure difficult, a task that has been attempted to reduce the compound's associated toxicity.[9]
Target of Bafilomycin
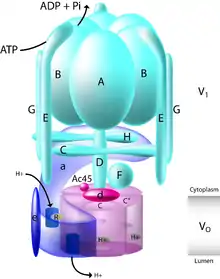
Within the cell, bafilomycin A1 specifically interacts with the proton pump V-ATPase. This large protein depends on Adenosine triphosphate (ATP) hydrolysis to pump protons across a biological membrane.[3] When bafilomycin and other inhibitors of V-ATPase, such as concanamycin, were first discovered in the 1980s they were used to establish the presence of V-ATPase in specialized cells types and tissues, characterizing the proton pump's distribution.[12] Structurally, V-ATPase consists of 13 distinct subunits that together make up the membrane spanning Vo and cytosolic V1 domains of the enzyme.[3] The V1 domain in the cytosol is made up of subunits A through H whereas the Vo domain is made up of subunits a, d, e, c, and c".[13]
V-ATPase Mechanism of Action
In order to move protons across the membrane, a proton first enters subunit a within the Vo domain through a cytoplasmic hemichannel. This allows conserved glutamic acid residues within the proteolipid ring of Vo subunits c and c" to become protonated. Adenosine triphosphate (ATP) is then hydrolyzed by the V1 domain of the enzyme, enabling both the rotation of the central stalk of the pump, made up of subunits D, F and d, and the rotation of the proteolipid ring. This rotation puts the protonated glutamic acid residues in contact with a luminal hemichannel located in subunit a. Within subunit a, arginine residues serve to stabilize the deprotonated form of glutamic acid and allow the release of their protons. This rotation and proton transfer brings the protons through the pump and across the membrane.[3][13]
Bafilomycin- V-ATPase Interaction
For more than ten years after bafilomycin was discovered as a V-ATPase inhibitor, the site of its interaction with V-ATPase was unclear. Beginning studies used the chromaffine granule V-ATPase to suggest that bafilomycin interacted with the Vo domain. Two further studies confirmed this hypothesis using V-ATPase from bovine clathrin coated vesicles. They showed that application of bafilomycin inhibited proton flow through Vo and that this inhibition could be overcome by adding back the Vo domain to the coated vesicles. Further narrowing bafilomycin's interaction site, they found that specific addition of just Vo subunit a could restore function. This suggested bafilomycin interacted specifically with subunit a of V-ATPase; however, another study contradicted this finding. A group found that by using a bafilomycin affinity chromatography column V-ATPase could be purified, and that addition of DCCD, an inhibitor of the Vo c subunit, drastically decreased bafilomycin's affinity for V-ATPase. This suggested that bafilomycin interacted more strongly with subunit c of the Vo domain. Two further studies then confirmed this interaction using radiolabeling and protein crystallization. It was further found, however, that amino acid changes within subunit a could also lower V-ATPase-Bafilomycin interaction, indicating a minor role of subunit a in bafilomycin binding in addition to subunit c.[12]
Overall, bafilomycin binds with nanomolar efficiency to the Vo c subunit of the V-ATPase complex.[3] Specifically, bafilomycin interacts with the proteolipid ring of Vo, inhibiting proton translocation.[13] Although the interaction between bafilomycin and V-ATPase is not covalent, its low dissociation constant of about 10 nM describes the strength of its interaction and can make the effects of bafilomycin difficult to reverse.[14]
V-ATPase Localization and Function
V-ATPase is ubiquitous in mammalian cells and plays an important role in many cellular processes. It is localized to the trans-golgi network and the cellular organelles that are derived from it, including lysosomes, secretory vesicles and endosomes.[15] V-ATPase can also be found within the plasma membrane. In mammals, location of the V-ATPase can be linked to the specific isoform of subunit a that the complex has. Isoforms a1 and a2 target V-ATPase intracellularly, to synaptic vesicles and endosomes respectively. Subunits a3 and a4, however, mediate V-ATPase localization to the plasma membrane in osteoclasts (a3) and renal intercalated cells (a4). If located at the lysosomal membrane, this results in the acidification of the lysosome as lumenal pH is lowered, enabling activity of lysosomal hydrolases. When V-ATPase is located at the plasma membrane, proton extrusion through the pump causes the acidification of the extracellular space, which is utilized by specialized cells such as osteoclasts, epididymal clear cells, and renal epithelial intercalated cells.[3]
Intracellular Function
As it promotes the acidification of lysosomes, endosomes, and secretory vesicles, V-ATPase contributes to processes including:
- vesicular/protein trafficking
- receptor recycling
- endocytosis
- protein degradation
- autophagy
- cell signaling
With its role in lysosomal acidification, V-ATPase is also crucial in driving the transport of ions and small molecules into the cytoplasm, particularly calcium and amino acids. Additionally, its acidification of endosomes is critical in receptor endocytosis as low pH tends to drive ligand release as well as receptor cleavage which contributes to signaling events, such as through the release of the intracellular domain of Notch.[3]
Plasma Membrane Function
When at the plasma membrane, V-ATPase function is critical in the acidification of the extracellular environment, which is seen with osteoclasts and epidiymal clear cells. When present at the plasma membrane in renal epithelial intercalated cells, V-ATPase is important for acid secretion, which contributes to the acidification of urine. In response to reduced plasma pH, increased levels of V-ATPase are typically trafficked to the plasma membrane in these cells by phosphorylation of the pump by Protein Kinase A (PKA).[3]
V-ATPase in Disease
Clinically, dysfunction of V-ATPase has been correlated with several diseases in humans. Some of these diseases include male infertility, osteopetrosis, and renal acidosis.[12] Additionally, V-ATPase can be found at the plasma membrane of some invasive cancer cells including breast, prostate and liver cancer, among others. In human lung cancer samples, V-ATPase expression was correlated with drug resistance.[13] A large number of V-ATPase subunit mutations have also been identified in a number of cancers, including follicular lymphomas.[3]
Cellular Action of Bafilomycin
As the target of Bafilomycin V-ATPase, is involved in many aspects of cellular function, Bafilomycin treatment greatly alters cellular processes.
Inhibition of Autophagy
Bafilomycin A1 is most known for its use as an autophagy inhibitor.[10][16] Autophagy is the process by which the cell degrades its own organelles and some proteins through the formation of autophagosomes. Autophagosomes then fuse with lysosomes facilitating the degradation of engulfed cargo by lysosomal proteases. This process is critical in maintaining the cell's store of amino acids and other nutrients during times of nutrient deprivation or other metabolic stresses.[17] Bafilomycin interferes with this process by inhibiting the acidification of the lysosome through its interaction with V-ATPase. Lack of lysosomal acidification prevents the activity of lysosomal proteases like cathepsins so that engulfed cargo can no longer be degraded.[16]
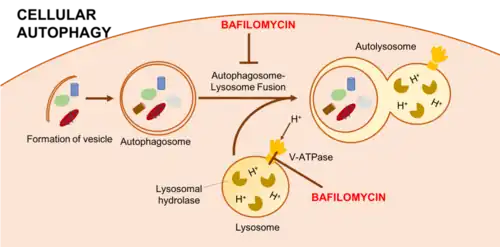
Since V-ATPase is widely distributed within the cell, Bafilomycin is only specific as an autophagy inhibitor for a short amount of time. Other effects are seen outside this short window, including interference in the trafficking of endosomes and proteasomal inhibition.[16]
In addition to blocking the acidification of the lysosome, Bafilomycin has been reported to block the fusion of autophagosomes with lysosomes.[10] This was initially found in a paper by Yamamoto, et al. in which the authors used bafilomycin A1 to treat rat hepatoma H-4-II-E cells. By electron microscopy, they saw a blockage of autophagosome-lysosome fusion after using bafilomycin at a concentration of 100 nM for 1 hour. This has been confirmed by other studies, particularly two that found decreased colocalization of mitochondria and lysosomes by fluorescence microscopy following a 12-24 hour treatment with 100 or 400 nM Bafilomycin. However, further studies have failed to see this inhibition of fusion with similar bafilomycin treatments. These contradictory results have been explained by time differences among treatments as well as use of different cell lines. The effect of Bafilomycin on autophagosome-lysosome fusion is complex and time dependent in each cell line.[8][18]
In neurons, an increase in the autophagosome marker LC3-II has been seen with Bafilomycin treatment. This occurs as autophagosomes fail to fuse with lysosomes, which normally stimulates the degradation of LC3-II.[19]
Induction of apoptosis
In PC12 cells, bafilomycin was found to induce apoptosis, or programmed cell death.[14] Additionally, in some cell lines it has been found to disrupt the electrochemical gradient of the mitochondria and induce the release of cytochrome c, which is an initiator of apoptosis.[8] Bafilomycin has also been shown to induce both inhibition of autophagy and subsequent induction of apoptosis in osteosarcoma cells[20] as well as other cancer cell lines.[3]
K+ transport
Bafilomycin acts as an ionophore, meaning it can transfer K+ ions across biological membranes.[12] Typically, the mitochondrial inner membrane is not permeable to K+ and maintains a set electrochemical gradient. In excitable cells, mitochondria can contain a K+ channel that, when opened, can cause mitochondrial stress by inducing mitochondrial swelling, changing the electrochemical gradient, and stimulating respiration. Bafilomycin A1 treatment can induce mitochondrial swelling in the presence of K+ ions, stimulate the oxidation of pyrimidine nucleotides and uncouple oxidative phosphorylation. Ascending concentrations of bafilomycin were found to linearly increase the amount of K+ that traversed the mitochondrial membrane, confirming it acts as an ionophore. Compared to other ionophores, however, bafilomycin has a low affinity for K+.[8]
Research Applications
Anti-tumorigenic
In many cancers, it has been found that various subunits of V-ATPase are upregulated.[13] Upregulation of these subunits appears to be correlated with increased tumor cell metastasis and reduced clinical outcome. Bafilomycin application has been shown to reduce cell growth in various cancer cell lines across multiple cancer types by induction of apoptosis. Additionally, in vitro bafilomycin's anti-proliferative effect appears to be specific to cancer cells over normal cells, which is seen with selective inhibition of hepatoblastoma cell growth compared to healthy hepatocytes.[3]
The mechanism by which bafilomycin causes this cancer specific anti-proliferative effect is multifactorial. In addition to the induction of caspase-dependent apoptosis through the mitochondrial pathway, bafilomycin also causes increased levels of reactive oxygen species and increased expression of HIF1alpha. These effects suggest that inhibition of V-ATPase with bafilomycin can induce a cellular stress response, including autophagy and eventual apoptosis. These somewhat contradictory effects of V-ATPase inhibition in terms of inhibition or induction of apoptosis demonstrate that bafilomycin's function is critically dependent on cellular context, and can mediate either a pro-survival or pro-death phenotype.[3][18]
In vivo bafilomycin reduced average tumor volume in MCF-7 and MDA-MB-231 xenograft mouse models by 50% and did not show toxic effects at a dosing of 1 mg/kg. Additionally, when combined with sorafenib, bafilomycin also caused tumor regression in MDA-MB-231 xenograft mice. In a HepG2 orthotropic HCC xenograft model in nude mice, bafilomycin prevented tumor growth.[3]
V-ATPase dysregulation is thought to play a role in resistance to cancer therapies, as aberrant acidification of the extracellular environment can protonate chemotherapeutics, preventing their entry into the cell.[3][13] It is unclear if` V-ATPase dysregulation is a direct cause of associated poor clinical outcome or if its dysregulation primarily effects the response to treatment. Although treatment with bafilomycin and cisplatin had a synergistic effect on cancer cell cytotoxicity.[3]
Anti-fungal
Bafilomycins have been shown to inhibit plasma membrane ATPase (P-ATPase) as well as the ATP-binding cassette (ABC) transporters. These transporters are identified as good anti-fungal targets as they render organisms unable to cope with cation stress.[7] When Cryptococcus neoformans was treated with bafilomycin, growth inhibition was observed.[21] Bafilomycin has also been used in C. neoformans in conjunction with calcineurin inhibitor FK506, displaying synergistic anti-fungal activity.[7]
Anti-parasitic
Bafilomycin has been shown to be active against Plasmodium falciparum, the causative agent of malaria. Upon infection of red blood cells, P. falciparum exports a membrane network into the red blood cell cytoplasm and also inserts several of its own proteins into the host membrane, including its own V-ATPase. This proton pump has a role in maintaining the intracellular pH of the infected red blood cell and facilitating the uptake of small metabolites at equilibrium. Treatment of the parasitized red blood cell with bafilomycin prevents the extracellular acidification, causing a dip in intracellular pH around the malarial parasite.[4][5]
Immunosuppressant
The inflammatory myopathy Inclusion Body Myositis (IBM) is relatively common in patients over 50 years of age and involves over activation of autophagic flux. In this condition, increased autophagy results in an increase in protein degradation and therefore an increase in the presentation of antigenic peptides in muscles. This can cause over-activation of immune cells. Treatment with bafilomycin can prevent the acidification of lysosomes and therefore autophagy, decreasing the number of antigenic peptides digested and displayed to the immune system.[6]
In Lupus patients, the autophagy pathway has been found to be altered in both B and T cells. Particularly, more autophagic vacuoles were seen in T cells as well as increased LC3-11 staining for autophagosomes, indicating increased autophagy. Increased autophagy can also be seen in naïve patient B cell subsets. Bafilomycin A1 treatment lowered the differentiation of plasmablasts and decreased their survival.[22]
Clearance of protein aggregates in neurodegenerative diseases
Neurodegenerative diseases typically display elevated levels of protein aggregates within the cell that contribute to dysfunction of neurons and eventual neuronal death. As a method of protein degradation within the cell, autophagy can traffic these protein aggregates to be degraded in the lysosome. Although it is unclear the exact role continuous autophagy, or autophagic flux, plays in neuronal homeostasis and disease states, it has been shown that autophagic dysfunction can be seen in neurodegenerative diseases.[19]
Bafilomycin is commonly used to study this autophagic flux in neurons, among other cell types. To do this, neurons are first put into nutrient rich conditions then into nutrient starved conditions to stimulate autophagy. Bafilomycin is co-administered in the condition of nutrient stress so that while autophagy is stimulated, bafilomycin blocks its final stage of autophagosome-lysosomal fusion resulting in the accumulation of autophagosomes. Levels of autophagy related proteins associated with autophagosomes, such as LC3, can then be monitored to determine the level of autophagosome formation induced by nutrient deprivation.[19]
In vitro Drug Interactions
Lysosomotropic Drugs
Some cationic drugs, such as chloroquine and sertraline, are known as lysosomotropic drugs. These drugs are weak bases that become protonated in the acidic environment of the lysosome. This traps the otherwise non-protonated compound within the lysosome, as protonation prevents its passage back across the lipid membrane of the organelle. This phenomenon is known as ion trapping. Trapping of the cationic compound also draws water into the lysosome through an osmotic effect, which can sometimes lead to vacuolization seen in in vitro cultured cells.[15][23]
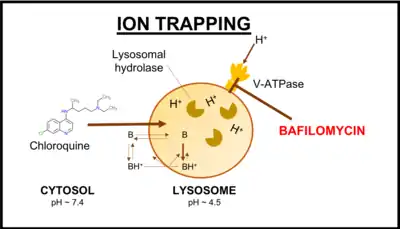
When one of these drugs is co-applied to cells with bafilomycin A1, the action of bafilomycin A1 prevents the acidification of the lysosome, therefore preventing the phenomenon of ion trapping in this compartment.[23] As the lysosome cannot acidify, lysosomotropic drugs do not become protonated and subsequently trapped in the lysosome in the presence of bafilomycin. Additionally, when cells are preloaded with lysosomotropic drugs in vitro, then treated with bafilomycin, bafilomycin acts to release the cationic compound from its accumulation in the lysosome.[15]
Pretreating cells with bafilomycin before administration of a cationic drug can alter the kinetics of the cationic compound. In a rabbit contractility assay, bafilomycin was used to pre-treat isolated rabbit aorta. The lipophilic agent xylometazoline, an alpha-adrenoreceptor agonist, displayed an increased effect when administered after bafilomycin treatment. With bafilomycin, faster contraction and relaxation of the aorta was seen as bafilomycin prevented the ion trapping of xylometazoline in the lysosome. Without pre-treatment with bafilomycin, the functional V-ATPase causes the lysosome to become a reservoir for xylometazoline, slowing its effect on contractility.[15]
Chloroquine
As a lysosomotroic drug, chloroquine typically accumulates in the lysosome disrupting their degradative function, inhibiting autophagy, and inducing apoptosis through Bax-dependent mechanisms. However, in cultured cerebellar granule neurons (CGNs) low treatment with Bafillomycin of 1 nM decreased chloroquine induced apoptosis without affecting chloroquine inhibition of autophagy. The exact mechanism of this protection is unknown, although it is hypothesized to lie downstream of autophagosome-lysosome fusion yet upstream of Bax induction of apoptosis.[10]
Chemotherapeutics
Bafilomycin has been shown to potentiate the effect of taxol in decreasing Matrix Metalloprotease (MMP) levels by depressing Bcl-xL's mitochondrial protective role. Additionally, within cisplatin resistant cells, V-ATPase expression was found to be increased, and co-treatment of bafilomycin with cisplatin sensitized these cells to cisplatin-induced cytotoxicity.[3] Bafilomycin has also been shown to increase the efficacy of EGFR inhibitors in anti-cancer applications.[24]
References
- ↑ Bafilomycin A1 product page from Fermentek
- 1 2 3 Dröse S, Altendorf K (January 1997). "Bafilomycins and concanamycins as inhibitors of V-ATPases and P-ATPases". The Journal of Experimental Biology. 200 (Pt 1): 1–8. PMID 9023991.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 Whitton B, Okamoto H, Packham G, Crabb SJ (August 2018). "Vacuolar ATPase as a potential therapeutic target and mediator of treatment resistance in cancer". Cancer Medicine. 7 (8): 3800–3811. doi:10.1002/cam4.1594. PMC 6089187. PMID 29926527.
- 1 2 Hayashi M, Yamada H, Mitamura T, Horii T, Yamamoto A, Moriyama Y (November 2000). "Vacuolar H(+)-ATPase localized in plasma membranes of malaria parasite cells, Plasmodium falciparum, is involved in regional acidification of parasitized erythrocytes". The Journal of Biological Chemistry. 275 (44): 34353–8. doi:10.1074/jbc.M003323200. PMID 10915784.
- 1 2 Marchesini N, Vieira M, Luo S, Moreno SN, Docampo R (November 2005). "A malaria parasite-encoded vacuolar H(+)-ATPase is targeted to the host erythrocyte". The Journal of Biological Chemistry. 280 (44): 36841–7. doi:10.1074/jbc.M507727200. PMID 16135514.
- 1 2 Keller CW, Schmidt J, Lünemann JD (June 2017). "Immune and myodegenerative pathomechanisms in inclusion body myositis". Annals of Clinical and Translational Neurology. 4 (6): 422–445. doi:10.1002/acn3.419. PMC 5454400. PMID 28589170.
- 1 2 3 Mayers D (2008). Antimicrobial drug resistance handbook. Volume 2, Clinical and epidemiological aspects. Totowa, N.J.: Humana. ISBN 9781603275958. OCLC 437345683.
- 1 2 3 4 Saris NE, Andersson MA, Mikkola R, Andersson LC, Teplova VV, Grigoriev PA, Salkinoja-Salonen MS (August 2009). "Microbial toxin's effect on mitochondrial survival by increasing K+ uptake". Toxicology and Industrial Health. 25 (7): 441–6. doi:10.1177/0748233709103405. PMID 19736254. S2CID 30966042.
- 1 2 3 4 Li Z, Du L, Zhang W, Zhang X, Jiang Y, Liu K, Men P, Xu H, Fortman JL, Sherman DH, Yu B, Gao S, Li S (April 2017). "Streptomyces lohii". The Journal of Biological Chemistry. 292 (17): 7095–7104. doi:10.1074/jbc.M116.751255. PMC 5409476. PMID 28292933.
- 1 2 3 4 Shacka JJ, Klocke BJ, Roth KA (2006). "Autophagy, bafilomycin and cell death: the "a-B-cs" of plecomacrolide-induced neuroprotection". Autophagy. 2 (3): 228–30. doi:10.4161/auto.2703. PMID 16874105.
- 1 2 Yu Z, Zhao LX, Jiang CL, Duan Y, Wong L, Carver KC, Schuler LA, Shen B (January 2011). "Bafilomycins produced by an endophytic actinomycete Streptomyces sp. YIM56209". The Journal of Antibiotics. 64 (1): 159–62. doi:10.1038/ja.2010.147. PMC 5592157. PMID 21102599.
- 1 2 3 4 Huss M, Wieczorek H (February 2009). "Inhibitors of V-ATPases: old and new players". The Journal of Experimental Biology. 212 (Pt 3): 341–6. doi:10.1242/jeb.024067. PMID 19151208.
- 1 2 3 4 5 6 Cotter K, Stransky L, McGuire C, Forgac M (October 2015). "Recent Insights into the Structure, Regulation, and Function of the V-ATPases". Trends in Biochemical Sciences. 40 (10): 611–622. doi:10.1016/j.tibs.2015.08.005. PMC 4589219. PMID 26410601.
- 1 2 Weisz OA (2003-01-01). "Acidification and protein traffic". International Review of Cytology. 226: 259–319. doi:10.1016/S0074-7696(03)01005-2. ISBN 9780123646309. PMID 12921239.
- 1 2 3 4 Marceau F, Bawolak MT, Lodge R, Bouthillier J, Gagné-Henley A, Gaudreault RC, Morissette G (February 2012). "Cation trapping by cellular acidic compartments: beyond the concept of lysosomotropic drugs". Toxicology and Applied Pharmacology. 259 (1): 1–12. doi:10.1016/j.taap.2011.12.004. hdl:20.500.11794/15930. PMID 22198553.
- 1 2 3 Vinod V, Padmakrishnan CJ, Vijayan B, Gopala S (April 2014). "'How can I halt thee?' The puzzles involved in autophagic inhibition". Pharmacological Research. 82: 1–8. doi:10.1016/j.phrs.2014.03.005. PMID 24657238.
- ↑ Duffy A, Le J, Sausville E, Emadi A (March 2015). "Autophagy modulation: a target for cancer treatment development". Cancer Chemotherapy and Pharmacology. 75 (3): 439–47. doi:10.1007/s00280-014-2637-z. PMID 25422156. S2CID 24642257.
- 1 2 Klionsky DJ, Elazar Z, Seglen PO, Rubinsztein DC (October 2008). "Does bafilomycin A1 block the fusion of autophagosomes with lysosomes?". Autophagy. 4 (7): 849–50. doi:10.4161/auto.6845. PMID 18758232.
- 1 2 3 Lumkwana D, du Toit A, Kinnear C, Loos B (June 2017). "Autophagic flux control in neurodegeneration: Progress and precision targeting-Where do we stand?". Progress in Neurobiology. 153: 64–85. doi:10.1016/j.pneurobio.2017.03.006. PMID 28385648. S2CID 3811723.
- ↑ Xie Z, Xie Y, Xu Y, Zhou H, Xu W, Dong Q (August 2014). "Bafilomycin A1 inhibits autophagy and induces apoptosis in MG63 osteosarcoma cells". Molecular Medicine Reports. 10 (2): 1103–7. doi:10.3892/mmr.2014.2281. PMID 24890793.
- ↑ Weber SM, Levitz SM, Harrison TS (August 2000). "Chloroquine and the fungal phagosome". Current Opinion in Microbiology. 3 (4): 349–53. doi:10.1016/S1369-5274(00)00102-8. PMID 10972492.
- ↑ Muller S, Brun S, René F, de Sèze J, Loeffler JP, Jeltsch-David H (August 2017). "Autophagy in neuroinflammatory diseases". Autoimmunity Reviews. 16 (8): 856–874. doi:10.1016/j.autrev.2017.05.015. PMID 28572049.
- 1 2 Kuzu OF, Toprak M, Noory MA, Robertson GP (March 2017). "Effect of lysosomotropic molecules on cellular homeostasis". Pharmacological Research. 117: 177–184. doi:10.1016/j.phrs.2016.12.021. PMID 28025106. S2CID 207368923.
- ↑ Ravanan P, Srikumar IF, Talwar P (November 2017). "Autophagy: The spotlight for cellular stress responses". Life Sciences. 188: 53–67. doi:10.1016/j.lfs.2017.08.029. PMID 28866100.