Beta-mannosidosis
| Beta-mannosidosis | |
|---|---|
| Other names: Beta-D-mannosidosis,Beta-mannosidase deficiency,Lysosomal beta A mannosidosis,Lysosomal beta-mannosidase deficiency[1] | |
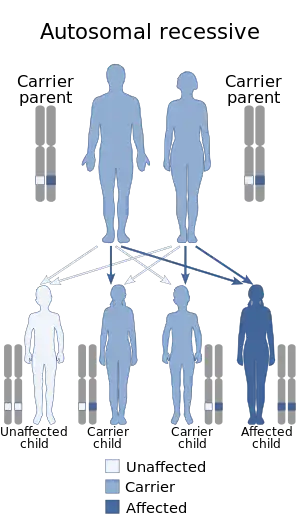 | |
| This condition is autosomal recessive in inheritance | |
| Symptoms | Respiratory infections, Hearing loss and Intellectual disability.[2] |
| Causes | Mutations in the MANBA gene[1] |
| Diagnostic method | Urine test[3] |
| Treatment | Based on symptoms[4] |
Beta-mannosidosis, also called lysosomal beta-mannosidase deficiency,[5] is a disorder of oligosaccharide metabolism caused by decreased activity of the enzyme beta-mannosidase. This enzyme is coded for by the gene MANBA, located at 4q22-25. Beta-mannosidosis is inherited in an autosomal recessive manner.[5]
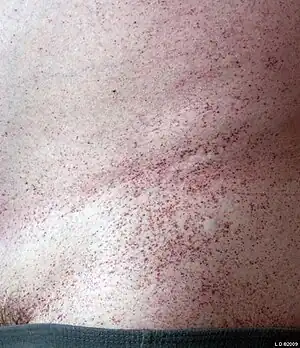
Affected individuals appear normal at birth, and can have a variable clinical presentation. Infantile onset forms show severe neurodegeneration, while some children have intellectual disability. Hearing loss and angiokeratomas are common features of the disease.[3][1]
Symptoms and signs
The initial affected individual described in 1986 had a complex phenotype, and was later found to have both beta-mannosidosis and Sanfilippo syndrome.[5]
People have been described with a wide spectrum of clinical presentations from infants and children with intellectual disability to adults who present with isolated skin findings (angiokeratomas).[5]
Most cases are identified in the first year of life with respiratory infections, hearing loss and intellectual disability. Because of its rarity, and non-specific clinical findings, beta-mannosidosis can go undiagnosed until adulthood, where it can present with intellectual disability and behavioral problems, including aggression.[6][2]
Cause
In terms of causation several mutations in the MANBA gene is the cause of beta-mannosidosis. The cytogenetic location of the gene is 4q24, furthermore the condition is inherited in an autosomal recessive manner[7][1][8]
Mechanism

The pathophysiology of this condition, is better comprehended, if one first looks at the normal function of beta-mannosidase such as its function of breaking down disaccharides[9][10]
Beta-mannosidase function is consistent with, it being a lysosomal enzyme catalyzing and thus involved in degradation route for N-linked oligosaccharide moieties(glycoproteins)[8]
Diagnosis
A diagnosis of beta-mannosidosis is suspected based on the following:[3]
- Presentation
- Urine testing (identify abnormal oligosaccharides)
- Enzymatic analysis
- Molecular testing
Differential diagnosis
Diagnostic techniques for this condition can be done to offer a DDx, via lectin histochemistry to distinguish between α-mannosidosis and beta-mannosidosis.[11]
Treatment
In terms of beta-mannosidosis treatment, there is none currently, individuals that exhibit muscle weakness or seizures are treated based on the symptoms, since there's no cure[4]
Frequency
About 20 individuals have been identified worldwide with Beta-mannosidosis.[12]
See also
- Beta-mannosidase
- Alpha-mannosidosis
References
- 1 2 3 4 Reference, Genetics Home. "beta-mannosidosis". Genetics Home Reference. Archived from the original on 2017-07-28. Retrieved 2017-07-13.
- 1 2 "Mannosidosis, beta A, lysosomal | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Archived from the original on 2018-01-02. Retrieved 2017-07-13.
- 1 2 3 Enns, Gregory M.; Steiner, Robert D.; Cowan, Tina M. (2009). "Lysosomal Disorders". In Sarafoglou, Kiriakie; Hoffmann, Georg F.; Roth, Karl S. (eds.). Pediatric Endocrinology and Inborn Errors of Metabolism (1st ed.). New York: McGraw-Hill Medical. pp. 721–755. ISBN 978-0-07-143915-2.
- 1 2 Kelly, Evelyn B. (2013). Encyclopedia of Human Genetics and Disease. ABC-CLIO. p. 514. ISBN 9780313387135. Archived from the original on 6 November 2021. Retrieved 10 December 2017.
- 1 2 3 4 Online Mendelian Inheritance in Man (OMIM): 248510
- ↑ Sedel, F.; Baumann, N.; Turpin, J. -C.; Lyon-Caen, O.; Saudubray, J. -M.; Cohen, D. (2007). "Psychiatric manifestations revealing inborn errors of metabolism in adolescents and adults". Journal of Inherited Metabolic Disease. 30 (5): 631–641. doi:10.1007/s10545-007-0661-4. PMID 17694356. S2CID 8419283.subscription required
- ↑ Reference, Genetics Home. "MANBA gene". Genetics Home Reference. Archived from the original on 2017-10-26. Retrieved 2017-10-25.
- 1 2 "OMIM Entry - * 609489 - MANNOSIDASE, BETA A, LYSOSOMAL; MANBA". www.omim.org. Archived from the original on 21 August 2019. Retrieved 8 November 2021.
- ↑ "MANBA mannosidase beta [Homo sapiens (human)] - Gene - NCBI". www.ncbi.nlm.nih.gov. Archived from the original on 8 November 2021. Retrieved 7 November 2021.
- ↑ Stütz, Arnold E.; Wrodnigg, Tanja M. (1 January 2016). "Chapter Four - Carbohydrate-Processing Enzymes of the Lysosome: Diseases Caused by Misfolded Mutants and Sugar Mimetics as Correcting Pharmacological Chaperones". Advances in Carbohydrate Chemistry and Biochemistry. Academic Press. pp. 225–302. Archived from the original on 10 November 2021. Retrieved 9 November 2021.
- ↑ Johnson, William (2015). Rosenberg's Molecular and Genetic Basis of Neurological and Psychiatric Disease (Fifth ed.). Academic Press. pp. 369–383. ISBN 978-0-12-410529-4.
- ↑ Syed Haneef, S. A.; George Priya Doss, C. (1 January 2016). "Chapter Eight - Personalized Pharmacoperones for Lysosomal Storage Disorder: Approach for Next-Generation Treatment". Advances in Protein Chemistry and Structural Biology. Academic Press. pp. 225–265. ISBN 978-0-12-804795-8. Archived from the original on 22 September 2021. Retrieved 8 November 2021.
Further reading
- Molho-Pessach, Vered; Bargal, Ruth; Abramowitz, Yigal; Doviner, Victoria; Ingber, Arieh; Raas-Rothschild, Annick; Ne'eman, Zvi; Zeigler, Marsha; Zlotogorski, Abraham (2007). "Angiokeratoma corporis diffusum in human beta-mannosidosis: Report of a new case and a novel mutation". Journal of the American Academy of Dermatology. 57 (3): 407–412. doi:10.1016/j.jaad.2007.01.037. ISSN 1097-6787. PMID 17420068.
- Huynh, T; Khan, JM; Ranganathan, S (30 November 2011). "A comparative structural bioinformatics analysis of inherited mutations in β-D-Mannosidase across multiple species reveals a genotype–phenotype correlation". BMC Genomics. 12 Suppl 3: S22. doi:10.1186/1471-2164-12-S3-S22. ISSN 1471-2164. PMC 3333182. PMID 22369051.
External links
| Classification | |
|---|---|
| External resources |
|