Sly syndrome
| Sly syndrome | |
|---|---|
| Other names: β-glucuronidase deficiency, β-glucuronidase deficiency mucopolysaccharidosis, | |
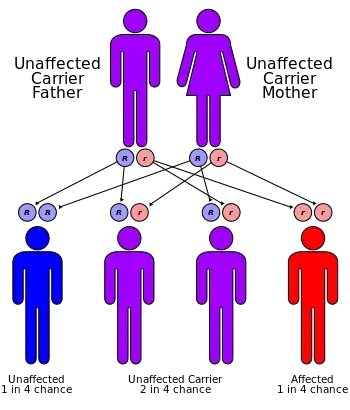 | |
| Sly syndrome has an autosomal recessive pattern of inheritance | |
Sly syndrome, also called mucopolysaccharidosis type VII (MPS-VII), is an autosomal recessive lysosomal storage disease caused by a deficiency of the enzyme β-glucuronidase. This enzyme is responsible for breaking down large sugar molecules called glycosaminoglycans (AKA GAGs, or mucopolysaccharides). The inability to break down GAGs leads to a buildup in many tissues and organs of the body. The severity of the disease can vary widely.[1]
Signs and symptoms
The most severe cases of Sly syndrome can result in hydrops fetalis, which results in fetal death or death soon after birth. Some people with Sly syndrome may begin to have symptoms in early childhood. Symptoms can include an enlarged head, fluid buildup in the brain, coarse facial features, enlarged tongue, enlarged liver, enlarged spleen, problems with the heart valves, and abdominal hernias. People with Sly syndrome may also suffer from sleep apnea, frequent lung infections, and problems with vision secondary to cloudy corneas. Sly syndrome causes various musculoskeletal abnormalities that worsen with age. These can include short stature, joint deformities, dysostosis multiplex, spinal stenosis, and carpal tunnel syndrome.[1]
While some individuals have developmental delay, others may have normal intelligence.[1] However, the accumulation of GAGs in the brain usually leads to the slowing of development from ages 1–3, and then a loss of previously learned skills until death.[2]
Genetics
The defective gene responsible for Sly syndrome is located on chromosome 7.[3]
Diagnosis

Most people with Sly disease will have elevated levels of GAGs seen in the urine. A confirmatory test is necessary for diagnosis. Skin cells and red blood cells of affected people will have low levels of β-glucuronidase activity. Sly syndrome can also be diagnosed through prenatal testing.[2]
Treatment
Vestronidase alfa-vjbk (trade name Mepsevii), an enzyme replacement therapy which is a recombinant form of human β-glucuronidase, is approved by U.S. Food and Drug Administration for the treatment of Sly syndrome.[4] Hematopoietic stem cell transplant (HSCT) has been used to treat other types of MPS diseases, but this is not yet available for MPS-VII. Animal experiments suggest that HSCT may be an effective treatment for MPS-VII in humans.[2]
Prognosis
The life expectancy of individuals with MPS VII varies depending on the symptoms. Some individuals are stillborn, while some may survive into adulthood.[1]
Epidemiology
MPS-VII is one of the rarest forms of MPS. It occurs in less than 1 in 250,000 births. As a family, MPS diseases occur in 1 in 25,000 births, and the larger family of lysosomal storage diseases occur in 1 out of 7,000 to 8,000 births.[2]
History
Sly syndrome was originally discovered in 1972.[2] It was named after its discoverer William S. Sly, an American biochemist who has spent nearly his entire academic career at Saint Louis University.[5][6]
References
- 1 2 3 4 "Mucopolysaccharidosis type VII". United States National Library of Medicine. 25 June 2019. Archived from the original on 3 July 2019. Retrieved 2 July 2019.
- 1 2 3 4 5 "A Guide to Understanding MPS VII" (PDF). National MPS Society. Archived (PDF) from the original on 3 July 2019. Retrieved 2 July 2019.
- ↑ Allanson, JE; Gemmill, RM; Hecht, BK; Johnsen, S; Wenger, DA (1988). "Deletion mapping of the beta-glucuronidase gene". American Journal of Medical Genetics. 29 (3): 517–522. doi:10.1002/ajmg.1320290307. PMID 3376995.
- ↑ McCafferty, EH; Scott, LJ (April 2019). "Vestronidase alfa: A review in mucopolysaccharidosis VII". BioDrugs. 33 (2): 233–240. doi:10.1007/s40259-019-00344-7. PMC 6469592. PMID 30848434.
- ↑ "slu.edu". Archived from the original on 2007-09-11. Retrieved 2007-12-31.
- ↑ Sly WS, Quinton BA, McAlister WH, Rimoin DL (1973). "Beta glucuronidase deficiency: report of clinical, radiologic, and biochemical features of a new mucopolysaccharidosis". J. Pediatr. 82 (2): 249–57. doi:10.1016/S0022-3476(73)80162-3. PMID 4265197.
External links
| Classification | |
|---|---|
| External resources |