Validation (drug manufacture)
Validation is the process of establishing documentary evidence demonstrating that a procedure, process, or activity carried out in testing and then production maintains the desired level of compliance at all stages. In the pharmaceutical industry, it is very important that in addition to final testing and compliance of products, it is also assured that the process will consistently produce the expected results.[1] The desired results are established in terms of specifications for outcome of the process. Qualification of systems and equipment is therefore a part of the process of validation. Validation is a requirement of food, drug and pharmaceutical regulating agencies such as the US FDA and their good manufacturing practices guidelines. Since a wide variety of procedures, processes, and activities need to be validated, the field of validation is divided into a number of subsections including the following:
- Equipment validation
- Facilities validation
- HVAC system validation
- Cleaning validation
- Process Validation
- Analytical method validation
- Computer system validation
Similarly, the activity of qualifying systems and equipment is divided into a number of subsections including the following:
- Design qualification (DQ)
- Component qualification (CQ)
- Installation qualification (IQ)
- Operational qualification (OQ)
- Performance qualification (PQ)
History
The concept of validation was first proposed by two Food and Drug Administration (FDA) officials, Ted Byers and Bud Loftus, in 1979 in USA, to improve the quality of pharmaceuticals.[2] It was proposed in direct response to several problems in the sterility of large volume parenteral market. The first validation activities were focused on the processes involved in making these products, but quickly spread to associated processes including environmental control, media fill, equipment sanitization and purified water production.
The concept of validation was first developed for equipment and processes and derived from the engineering practices used in delivery of large pieces of equipment that would be manufactured, tested, delivered and accepted according to a contract[3] The use of validation spread to other areas of industry after several large-scale problems highlighted the potential risks in the design of products. The most notable is the Therac-25 incident.[4] Here, the software for a large radiotherapy device was poorly designed and tested. In use, several interconnected problems led to several devices giving doses of radiation several thousands of times higher than intended, which resulted in the death of three patients and several more being permanently injured.
In 2005 an individual wrote a standard by which the transportation process could be validated for cold chain products. This standard was written for a biological manufacturing company and was then written into the PDA's Technical Report # 39,thus establishing the industry standard for cold chain validation. This was critical for the industry due to the sensitivity of drug substances, biologics and vaccines to various temperature conditions. The FDA has also been very focused on this final area of distribution and the potential for a drug substances quality to be impacted by extreme temperature exposure. 4.6. Accuracy: Accuracy of an analytical procedure is the closeness of test results obtained by that procedure to the true value. The accuracy of an analytical procedure shall be established across its range. 4.7. Precision: The precision of an analytical procedure expresses the closeness of agreement between a series of measurements obtained from multiple sampling of the same homogeneous sample under the prescribed conditions. 4.8. Method precision (Repeatability): Method precision carried out on different test preparation of a homogenous sample within short interval of time under same experimental conditions. 4.9. Intermediate precision (Ruggedness): Intermediate precision (Ruggedness) expresses within-laboratories variations i.e. different days, different analysts, different equipment etc. 4.10. Range: The range of an analytical procedure is the interval between the upper and lower concentration of analyte in the sample for which it has been demonstrated that the analytical procedure has a suitable level of precision, accuracy and linearity
Reasons for validation
FDA, or any other food and drugs regulatory agency around the globe not only ask for a product that meets its specification but also require a process, procedures, intermediate stages of inspections, and testing adopted during manufacturing are designed such that when they are adopted they produce consistently similar, reproducible, desired results which meet the quality standard of product being manufactured and complies the Regulatory and Security Aspects. Such procedures are developed through the process of validation. This is to maintain and assure a higher degree of quality of food and drug products. "Process validation is defined as the collection and evaluation of data, from the process design stage through commercial production, which establishes scientific evidence that a process is capable of consistently delivering quality product. Process validation involves a series of activities taking place over the lifecycle of the product and process.".[5] A properly designed system will provide a high degree of assurance that every step, process, and change has been properly evaluated before its implementation. Testing a sample of a final product is not considered sufficient evidence that every product within a batch meets the required specification.
Validation Master Plan
The Validation Master Plan is a document that describes how and when the validation program will be executed in a facility. Even though it is not mandatory, it is the document that outlines the principles involved in the qualification of a facility, defines the areas and systems to be validated and provides a written program for achieving and maintaining a qualified facility with validated processes. It is the foundation for the validation program and should include process validation, facility and utility qualification and validation, equipment qualification, cleaning and computer validation. The regulations also set out an expectation that the different parts of the production process are well defined and controlled, such that the results of that production will not substantially change over time.
The validation process
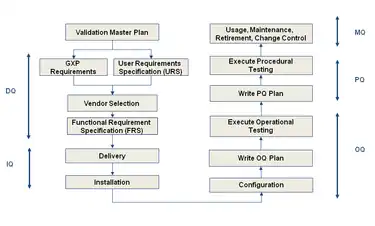
The validation scope, boundaries and responsibilities for each process or groups of similar processes or similar equipment's must be documented and approved in a validation plan. These documents, terms and references for the protocol authors are for use in setting the scope of their protocols. It must be based on a Validation Risk Assessment (VRA) to ensure that the scope of validation being authorised is appropriate for the complexity and importance of the equipment or process under validation. Within the references given in the VP the protocol authors must ensure that all aspects of the process or equipment under qualification; that may affect the efficacy, quality and or records of the product are properly qualified. Qualification includes the following steps:
- Design qualification (DQ)- Demonstrates that the proposed design (or the existing design for an off-the-shelf item) will satisfy all the requirements that are defined and detailed in the User Requirements Specification (URS). Satisfactory execution of the DQ is a mandatory requirement before construction (or procurement) of the new design can be authorised.
- Installation qualification (IQ) – Demonstrates that the process or equipment meets all specifications, is installed correctly, and all required components and documentation needed for continued operation are installed and in place.
- Operational qualification (OQ) – Demonstrates that all facets of the process or equipment are operating correctly.
- Performance qualification (PQ) – Demonstrates that the process or equipment performs as intended in a consistent manner over time.
- Component qualification (CQ) – is a relatively new term developed in 2005. This term refers to the manufacturing of auxiliary components to ensure that they are manufactured to the correct design criteria. This could include packaging components such as folding cartons, shipping cases, labels or even phase change material. All of these components must have some type of random inspection to ensure that the third party manufacturer's process is consistently producing components that are used in the world of GMP at drug or biologic manufacturer.
There are instances when it is more expedient and efficient to transfer some tests or inspections from the IQ to the OQ, or from the OQ to the PQ. This is allowed for in the regulations, provided that a clear and approved justification is documented in the Validation Plan (VP).
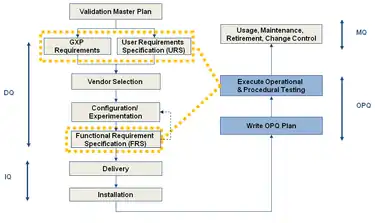
This combined testing of OQ and PQ phases is sanctioned by the European Commission Enterprise Directorate-General within ‘Annex 15 to the EU Guide to Good Manufacturing Practice guide’ (2001, p. 6) which states that:
"Although PQ is described as a separate activity, it may in some cases be appropriate to perform it in conjunction with OQ."
Computer System Validation
This requirement has naturally expanded to encompass computer systems used both in the development and production of, and as a part of pharmaceutical products, medical devices, food, blood establishments, tissue establishments, and clinical trials. In 1983 the FDA published a guide to the inspection of Computerized Systems in Pharmaceutical Processing, also known as the 'bluebook'.[6] Recently both the American FDA and the UK Medicines and Healthcare products Regulatory Agency have added sections to the regulations specifically for the use of computer systems. In the UK, computer validation is covered in Annex 11 of the EU GMP regulations (EMEA 2011). The FDA introduced 21 CFR Part 11 for rules on the use of electronic records, electronic signatures (FDA 1997). The FDA regulation is harmonized with ISO 8402:1994,[7] which treats "verification" and "validation" as separate and distinct terms. On the other hand, many software engineering journal articles and textbooks use the terms "verification" and "validation" interchangeably, or in some cases refer to software "verification, validation, and testing (VV&T)" as if it is a single concept, with no distinction among the three terms. The General Principles of Software Validation (FDA 2002) defines verification as "Software verification provides objective evidence that the design outputs of a particular phase of the software development life cycle meet all of the specified requirements for that phase."[8] It also defines Validation as "Confirmation by examination and provision of objective evidence that software specifications conform to user needs and intended uses, and that the particular requirements implemented through software can be consistently fulfilled". The software validation guideline states: “The software development process should be sufficiently well planned, controlled, and documented to detect and correct unexpected results from software changes." Annex 11 states "The validation documentation and reports should cover the relevant steps of the life cycle."
Weichel (2004) recently found that over twenty warning letters issued by the FDA to pharmaceutical companies specifically cited problems in Computer System Validation between 1997 and 2001.[9]
Probably the best known industry guidance available is the GAMP Guide, now in its fifth edition and known as GAMP5 published by ISPE (2008).[10] This guidance gives practical advice on how to satisfy regulatory requirements.
Scope of Computer Validation
The definition of validation above discusses production of evidence that a system will meet its specification. This definition does not refer to a computer application or a computer system but to a process. The main implications in this are that validation should cover all aspects of the process including the application, any hardware that the application uses, any interfaces to other systems, the users, training and documentation as well as the management of the system and the validation itself after the system is put into use. The PIC/S guideline (PIC/S 2004) defines this as a 'computer related system'.[11] Much effort is expended within the industry upon validation activities, and several journals are dedicated to both the process and methodology around validation, and the science behind it.[12][13][14][15]
Risk Based Approach To Computer Validation
In the recent years, a risk-based approach has been adopted within the industry, where the testing of computer systems (emphasis on finding problems) is wide-ranging and documented but not heavily evidenced (i.e. hundreds of screen prints are not gathered during testing). Annex 11 states "Risk management should be applied throughout the lifecycle of the computerised system taking into account patient safety, data integrity and product quality. As part of a risk management system, decisions on the extent of validation and data integrity controls should be based on a justified and documented risk assessment of the computerised system."
The subsequent validation or verification of computer systems targets only the "GxP critical" requirements of computer systems. Evidence (e.g. screen prints) is gathered to document the validation exercise. In this way it is assured that systems are thoroughly tested, and that validation and documentation of the "GxP critical" aspects is performed in a risk-based manner, optimizing effort and ensuring that computer system's fitness for purpose is demonstrated.
The overall risk posed by a computer system is now generally considered to be a function of system complexity, patient/product impact, and pedigree (Configurable-Off-The-Shelf or Custom-written for a certain purpose). A lower risk system should merit a less in-depth specification/testing/validation approach. (e.g. The documentation surrounding a spreadsheet containing a simple but "GxP" critical calculation should not match that of a Chromatography Data System with 20 Instruments)
Determination of a "GxP critical" requirement for a computer system is subjective, and the definition needs to be tailored to the organisation involved. However, in general a "GxP" requirement may be considered to be a requirement which leads to the development/configuration of a computer function which has a direct impact on patient safety, the pharmaceutical product being processed, or has been developed/configured to meet a regulatory requirement. In addition if a function has a direct impact on GxP data (security or integrity) it may be considered "GxP critical".
Product life cycle approach in validation
Validation process efforts must account for the complete product life cycle, including developmental procedures adapted for qualification of a drug product commencing with its research and development phase, rationale for adapting a best fit formula which represents the relationship between required outputs and specified inputs, and procedure for manufacturing. Each step is required to be justified and monitored in order to provide a good quality food and drug product. The FDA emphasizes the product life cycle approach in its evaluation of manufacturer regulatory compliance as well.
See also
References
- ↑ Validation definition and FDA, Regulatory agencies guidelines requirement Accessed 27 Feb 2014.
- ↑ Agalloco, J. (1995). "Validation: an unconventional review and reinvention". Validation: An Unconventional Review and Reinvention. 49 (4): 175–179.
- ↑ Hoffmann, A., Kahny-Simonius, J., Plattner, M., Schmidli-Vckovski, V., & Kronseder, C. (1998), 'Computer system validation: An overview of official requirements and standards', Pharmaceutica Acta Helvetiae, vol. 72, no. 6, pp. 317–325.
- ↑ Leveson, N. G. & Turner, C. S. (1993), 'An investigation of the Therac-25 accidents', Computer, vol. 26, no. 7, pp. 18–41.
- ↑ FDA (1987). "Guidelines on General Principles of Process Validation".
{{cite journal}}: Cite journal requires|journal=(help) - ↑ FDA (1983). "Guide to Inspection of Computerised Systems (The Blue Book)". US Food and Drug Administration, Maryland, USA.
- ↑ International Organization for Standardization, Geneva, Switzerland (1994). "ISO 8402:1994: Quality management and quality assurance—Vocabulary".
{{cite journal}}: Cite journal requires|journal=(help)CS1 maint: multiple names: authors list (link) - ↑ "General Principles of Software Validation; Final Guidance for Industry and FDA Staff" (PDF). US FDA. 2002. Retrieved February 27, 2013.
- ↑ Weichel, P. (2004). "Survey of Published FDA Warning Letters with Comment on Part 11 (21 CFR Part 11)". Journal of Validation Technology. 11 (1): 62–66.
- ↑ ISPE (2008). "GAMP5: Risk Based Approach to Computer Compliance". International Society for Pharmaceutical Engineers, Tampa, FL.
- ↑ PIC/S (2004). "Good Practices for Computerised Systems in Regulated "GXP" Environments, Report PI 011-2". Pharmaceutical Inspection Convention, Geneva.
- ↑ Smith, H. G. (2001). "Considerations for Improving Software Validation, Securing better assurance for less cost". Journal of Validation Technology. 7 (2): 150–157.
- ↑ Tracy, D. S. & Nash, R. A. (2002). "A Validation Approach for Laboratory Information Management Systems". Journal of Validation Technology. 9 (1): 6–14.
- ↑ Lucas, I. (2003). "Testing Times in Computer Validation". Journal of Validation Technology. 9 (2): 153–161.
- ↑ Balogh, M. & Corbin, V. (2005). "Taming the Regulatory Beast: Regulation vs Functionalism". Pharmaceutical Technology Europe. 17 (3): 55–58.
- Bibliography
- Health Canada Validation Guidelines
- Akers, J. (1993), 'Simplifying and improving Process Validation', Journal of Parenteral Science and Technology, vol. 47, no. 6, pp. 281–284.
- ASTM E2537 Guide for Application of Continuous Quality Verification for Pharmaceutical and Biopharmaceutical Manufacturing
- EMEA (1998), EUDRALEX Volume 4 – Medicinal Products for Human and Veterinary Use : Good Manufacturing Practice, European Medicines Agency, London
- European Commission Enterprise Directorate-General (2001), Final Version of Annex 15 to the EU Guide to Good Manufacturing Practice, Qualification and Validation, Brussels. European Commission Enterprise Directorate-General.
- US FDA: Guideline on general principles of Process Validation
- Part 11: Electronic Records; Electronic Signatures,Code of Federal Regulations
- Garston Smith, H. (2001), 'Considerations for Improving Software Validation', Journal of Validation Technology, vol. 7, no. 2, pp. 150–157.
- IT Pharma Validation Europe: News and Updates on Computer System Validation and Infrastructure Qualification – e.g. EudraLex Volume 4 – Annex 11 computerised systems – revision January 2011
- Lopez, Orlando (2002), “21 CFR Part 11 – A Complete Guide to International Compliance,” published by Sue Horwood Publishing Limited.
- McDowall, R. D. (2005), 'Effective and practical risk management options for computerised system validation', The Quality Assurance Journal, vol. 9, no. 3, pp. 196–227.
- Parker G, (2005) ‘Developing Appropriate Validation and Testing Strategies’ Presented for Scimcon Ltd at the Thermo Informatics World Conference. North America.
- Powell-Evans, K. (1998), 'Streamlining Validation', Pharmaceutical Technology Europe, vol. 10, no. 12, pp. 48–52.
- Segalstad, S.H (2008), ‘International IT Regulations and Compliance: Quality Standards in the Pharmaceutical and Regulated Industries, John Wiley & Sons , pp. 157 – 178.
- Swartz, M. (2006) ‘Analytical Instrument Qualification’, Avanstar [online], available at: http://www.advanstar.com/test/pharmascience/pha-sci_supp-promos/phasci_reg_guidance/articles/Instrumentation1_Swartz_rv.pdf (Accessed 29 March 2009).
- Validating Software used for the Pharmaceutical Industry. (2007). Retrieved July 6, 2009, from http://www.plainsite.net/validation/validation.htm
- WHO Technical Report Series, No. 937, 2006. Annex 4. Appendix 5. 2006
- Wingate, G.A.S. (2004), 'Computer Systems Validation: Quality Assurance, Risk Management, and Regulatory Compliance for the Pharmaceutical and Healthcare Industry', Interpharm Press.
- Guidance for Industry. Process Validation: General Principles and Practices. U.S. Department of Health and Human Services Food and Drug Administration. January 2011.