Adams–Oliver syndrome
| Adams–Oliver syndrome | |
|---|---|
| Other names: Congenital scalp defects with distal limb anomalies[1] | |
Adams–Oliver syndrome (AOS) is a rare congenital disorder characterized by defects of the scalp and cranium (cutis aplasia congenita), transverse defects of the limbs, and mottling of the skin.
Signs and symptoms
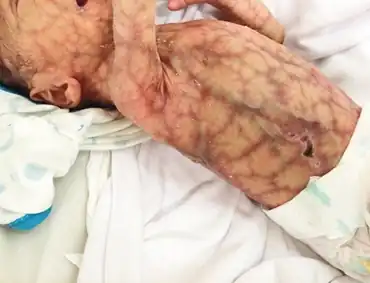
Two key features of AOS are aplasia cutis congenita with or without underlying bony defects and terminal transverse limb defects.[2] Cutis aplasia congenita is defined as missing skin over any area of the body at birth; in AOS skin aplasia occurs at the vertex of the skull. The size of the lesion is variable and may range from solitary round hairless patches to complete exposure of the cranial contents. There are also varying degrees of terminal limb defects (for example, shortened digits) of the upper extremities, lower extremities, or both. Individuals with AOS may have mild growth deficiency, with height in the low-normal percentiles. The skin is frequently observed to have a mottled appearance (cutis marmorata telangiectatica congenita). Other congenital anomalies, including cardiovascular malformations, cleft lip and/or palate, abnormal renal system, and neurologic disorders manifesting as seizure disorders and developmental delay are sometimes observed. Variable defects in blood vessels have been described, including hypoplastic aortic arch, middle cerebral artery, pulmonary arteries. Other vascular abnormalities described in AOS include absent portal vein, portal sclerosis, arteriovenous malformations, abnormal umbilical veins, and dilated renal veins.
Genetics
AOS was initially described as having autosomal dominant inheritance due to the reports of families with multiple affected family members in more than one generation.[3] The severity of the condition can vary between family members, suggestive of variable expressivity and reduced penetrance of the disease-causing allele. Subsequently, it was reported that some cases of AOS appear to have autosomal recessive inheritance, perhaps with somewhat more severe phenotypic effects.
Six AOS genes have been identified: ARHGAP31,[4] DOCK6,[5] RBPJ,[6] EOGT,[7][8] NOTCH1,[9][10] and DLL4.[11] ARHGAP31 and DOCK6 are both regulatory proteins that control members of the Rho family of GTPases and specifically regulate the activity of Cdc42 and Rac1. Autosomal dominant mutations in ARHGAP31 (a GTPase-activating protein) and autosomal recessive mutations in DOCK6 (a guanine nucleotide exchange factor) cause an accumulation of the inactive GTPase and lead to defects of the cytoskeleton.
RBPJ, EOGT, NOTCH1 and DLL4 are all involved in the Notch signalling pathway. Mutations in EOGT are found in AOS with autosomal recessive inheritance;[7] the other three genes account for cases with autosomal dominant inheritance.
Mechanism
The precise mechanism underlying the congenital abnormalities observed in AOS is unknown. Similar terminal transverse limb anomalies and cardiovascular malformations are seen in animal models of hypoxic insults during the first trimester.[12][13] Combined with the common association of cardiac and vascular abnormalities in AOS, it has been hypothesized that the spectrum of defects observed in AOS could be due to a disorder of vasculogenesis.
In rare cases, AOS can be associated with chromosomal translocations. A panel of candidate genes (including ALX4, ALX1, MSX1, MSX2, P63, RUNX2 and HOXD13) were tested but no disease-causing mutations were identified.[14][15] More recently, mutations in six genes have been identified, highlighting the Rho family of GTPases and the Notch signalling pathway as important factors in the pathogenesis of AOS.
Diagnosis
The diagnosis of AOS is a clinical diagnosis based on the specific features described above. A system of major and minor criteria was proposed.[16]
| Major features | Minor features |
|---|---|
| Terminal transverse limb defects | Cutis marmorata |
| Aplasia cutis congenita | Congenital heart defect |
| Family history of AOS | Vascular anomaly |
The combination of two major criteria would be sufficient for the diagnosis of AOS, while a combination of one major and one minor feature would be suggestive of AOS. Genetic testing can be performed to test for the presence of mutation in one of the known genes, but these so far only account for an estimated 50% of patients with AOS. A definitive diagnosis may therefore not be achieved in all cases.
Management
Management of AOS is largely symptomatic and aimed at treating the various congenital anomalies present in the individual. When the scalp and/or cranial bone defects are severe, early surgical intervention with grafting is indicated.
Prognosis
The overall prognosis is excellent in most cases. Most children with Adams–Oliver syndrome can likely expect to have a normal life span. However, individuals with more severe scalp and cranial defects may experience complications such as hemorrhage and meningitis, leading to long-term disability.
Epidemiology
AOS is a rare genetic disorder and the annual incidence or overall prevalence of AOS is unknown. Approximately 100 individuals with this disorder have been reported in the medical literature.
History
AOS was first reported by the American pediatric cardiologist Forrest H. Adams and the clinical geneticist Clarence Paul Oliver in a family with eight affected members.[3]
Citations
- ↑ RESERVED, INSERM US14-- ALL RIGHTS. "Orphanet: Adams Oliver syndrome". www.orpha.net. Archived from the original on 28 November 2017. Retrieved 16 May 2019.
- ↑ Mašek, Jan; Andersson, Emma R. (2017-05-15). "The developmental biology of genetic Notch disorders". Development. 144 (10): 1743–1763. doi:10.1242/dev.148007. ISSN 0950-1991. PMID 28512196.
- 1 2 Adams, Forrest H.; Oliver, C. P. (1945-01-01). "HEREDITARY DEFORMITIES IN MAN Due to Arrested Development". Journal of Heredity. 36 (1): 3–7. doi:10.1093/oxfordjournals.jhered.a105415. ISSN 0022-1503.
- ↑ Southgate, Laura; Machado, Rajiv D.; Snape, Katie M.; Primeau, Martin; Dafou, Dimitra; Ruddy, Deborah M.; Branney, Peter A.; Fisher, Malcolm; Lee, Grace J. (2011-05-13). "Gain-of-function mutations of ARHGAP31, a Cdc42/Rac1 GTPase regulator, cause syndromic cutis aplasia and limb anomalies". American Journal of Human Genetics. 88 (5): 574–585. doi:10.1016/j.ajhg.2011.04.013. ISSN 1537-6605. PMC 3146732. PMID 21565291.
- ↑ Shaheen, Ranad; Faqeih, Eissa; Sunker, Asma; Morsy, Heba; Al-Sheddi, Tarfa; Shamseldin, Hanan E.; Adly, Nouran; Hashem, Mais; Alkuraya, Fowzan S. (2011-08-12). "Recessive mutations in DOCK6, encoding the guanidine nucleotide exchange factor DOCK6, lead to abnormal actin cytoskeleton organization and Adams-Oliver syndrome". American Journal of Human Genetics. 89 (2): 328–333. doi:10.1016/j.ajhg.2011.07.009. ISSN 1537-6605. PMC 3155174. PMID 21820096.
- ↑ Hassed, Susan J.; Wiley, Graham B.; Wang, Shaofeng; Lee, Ji-Yun; Li, Shibo; Xu, Weihong; Zhao, Zhizhuang J.; Mulvihill, John J.; Robertson, James (2012-08-10). "RBPJ mutations identified in two families affected by Adams-Oliver syndrome". American Journal of Human Genetics. 91 (2): 391–395. doi:10.1016/j.ajhg.2012.07.005. ISSN 1537-6605. PMC 3415535. PMID 22883147.
- 1 2 Shaheen, Ranad; Aglan, Mona; Keppler-Noreuil, Kim; Faqeih, Eissa; Ansari, Shinu; Horton, Kim; Ashour, Adel; Zaki, Maha S.; Al-Zahrani, Fatema (2013-04-04). "Mutations in EOGT confirm the genetic heterogeneity of autosomal-recessive Adams-Oliver syndrome". American Journal of Human Genetics. 92 (4): 598–604. doi:10.1016/j.ajhg.2013.02.012. ISSN 1537-6605. PMC 3617382. PMID 23522784.
- ↑ Cohen, Idan; Silberstein, Eldad; Perez, Yonatan; Landau, Daniella; Elbedour, Khalil; Langer, Yshaia; Kadir, Rotem; Volodarsky, Michael; Sivan, Sara (2014-03-01). "Autosomal recessive Adams-Oliver syndrome caused by homozygous mutation in EOGT, encoding an EGF domain-specific O-GlcNAc transferase". European Journal of Human Genetics. 22 (3): 374–378. doi:10.1038/ejhg.2013.159. ISSN 1476-5438. PMC 3925282. PMID 23860037.
- ↑ Stittrich, Anna-Barbara; Lehman, Anna; Bodian, Dale L.; Ashworth, Justin; Zong, Zheyuan; Li, Hong; Lam, Patricia; Khromykh, Alina; Iyer, Ramaswamy K. (2014-09-04). "Mutations in NOTCH1 cause Adams-Oliver syndrome". American Journal of Human Genetics. 95 (3): 275–284. doi:10.1016/j.ajhg.2014.07.011. ISSN 1537-6605. PMC 4157158. PMID 25132448.
- ↑ Southgate, Laura; Sukalo, Maja; Karountzos, Anastasios S. V.; Taylor, Edward J.; Collinson, Claire S.; Ruddy, Deborah; Snape, Katie M.; Dallapiccola, Bruno; Tolmie, John L. (2015-08-01). "Haploinsufficiency of the NOTCH1 Receptor as a Cause of Adams-Oliver Syndrome With Variable Cardiac Anomalies". Circulation: Cardiovascular Genetics. 8 (4): 572–581. doi:10.1161/CIRCGENETICS.115.001086. ISSN 1942-3268. PMC 4545518. PMID 25963545.
- ↑ Meester, Josephina A. N.; Southgate, Laura; Stittrich, Anna-Barbara; Venselaar, Hanka; Beekmans, Sander J. A.; den Hollander, Nicolette; Bijlsma, Emilia K.; Helderman-van den Enden, Appolonia; Verheij, Joke B. G. M. (2015-09-03). "Heterozygous Loss-of-Function Mutations in DLL4 Cause Adams-Oliver Syndrome". American Journal of Human Genetics. 97 (3): 475–482. doi:10.1016/j.ajhg.2015.07.015. ISSN 1537-6605. PMC 4564989. PMID 26299364.
- ↑ Webster, William S.; Abela, Dominique (2007-09-01). "The effect of hypoxia in development". Birth Defects Research Part C: Embryo Today: Reviews. 81 (3): 215–228. doi:10.1002/bdrc.20102. ISSN 1542-975X. PMID 17963271.
- ↑ Ghatpande, Satish K.; Billington, Charles J.; Rivkees, Scott A.; Wendler, Christopher C. (2008-03-01). "Hypoxia induces cardiac malformations via A1 adenosine receptor activation in chicken embryos". Birth Defects Research. Part A, Clinical and Molecular Teratology. 82 (3): 121–130. doi:10.1002/bdra.20438. ISSN 1542-0760. PMC 3752680. PMID 18186126.
- ↑ Verdyck, Pieter; Holder-Espinasse, Muriel; Hul, Wim Van; Wuyts, Wim (2003-06-01). "Clinical and molecular analysis of nine families with Adams-Oliver syndrome". European Journal of Human Genetics. 11 (6): 457–463. doi:10.1038/sj.ejhg.5200980. ISSN 1018-4813. PMID 12774039.
- ↑ Verdyck, P.; Blaumeiser, B.; Holder-Espinasse, M.; Van Hul, W.; Wuyts, W. (2006-01-01). "Adams-Oliver syndrome: clinical description of a four-generation family and exclusion of five candidate genes". Clinical Genetics. 69 (1): 86–92. doi:10.1111/j.1399-0004.2006.00552.x. ISSN 0009-9163. PMID 16451141. S2CID 29732072.
- ↑ Snape, Katie M. G.; Ruddy, Deborah; Zenker, Martin; Wuyts, Wim; Whiteford, Margo; Johnson, Diana; Lam, Wayne; Trembath, Richard C. (2009-08-01). "The spectra of clinical phenotypes in aplasia cutis congenita and terminal transverse limb defects". American Journal of Medical Genetics Part A. 149A (8): 1860–1881. doi:10.1002/ajmg.a.32708. ISSN 1552-4833. PMID 19610107. S2CID 36061071.
References
- Jones, Kenneth L (1997). Smith's Recognizable Patterns of Human Malformation (5th ed.). Saunders. ISBN 0-7216-6115-7.
- James, William; Berger, Timothy; Elston, Dirk (2005). Andrews' Diseases of the Skin: Clinical Dermatology (10th ed.). Saunders. ISBN 0-7216-2921-0.
- Baskar S, Kulkarni ML, Kulkarni AM, Vittalrao S, Kulkarni PM (2009). "Adams–Oliver syndrome: Additions to the clinical features and possible role of BMP pathway". Am J Med Genet A. 149 (8): 1678–1684. doi:10.1002/ajmg.a.32938. PMID 19606482. S2CID 205311375.
- Bonafede RP, Beighton P (1979). "Autosomal dominant inheritance of scalp defects with ectrodactyly". Am J Med Genet. 3 (1): 35–41. doi:10.1002/ajmg.1320030109. PMID 474617.
- Maniscalco M, Zedda A, Faraone S, de Laurentiis G, Verde R, Molese V, Lapiccirella G, Sofia M (2005). "Association of Adams–Oliver syndrome with pulmonary arterio-venous malformation in the same family: a further support to the vascular hypothesis". Am J Med Genet A. 136 (3): 269–274. doi:10.1002/ajmg.a.30828. PMID 15948197. S2CID 3093562.
- McGoey RR, Lacassie Y (2008). "Adams–Oliver syndrome in siblings with central nervous system findings, epilepsy, and developmental delay: refining the features of a severe autosomal recessive variant". Am J Med Genet A. 146 (4): 488–491. doi:10.1002/ajmg.a.32163. PMID 18203152. S2CID 205308934.
- Whitley CB, Gorlin RJ (1991). "Adams–Oliver syndrome revisited". Am J Med Genet. 40 (3): 319–326. doi:10.1002/ajmg.1320400315. PMID 1951437.
- Zapata HH, Sletten LJ, Pierpont ME (1995). "Congenital cardiac malformations in Adams–Oliver syndrome". Clin Genet. 47 (2): 80–84. doi:10.1111/j.1399-0004.1995.tb03928.x. PMID 7606848. S2CID 13643644.
External links
| Classification | |
|---|---|
| External resources |
|