Angiofibroma of soft tissue
| Angiofibroma of soft tissue | |
|---|---|
| Specialty | Dermatology, General surgery, Pathology |
| Types | Benign |
| Causes | unknown |
| Treatment | Surgical removal |
| Prognosis | Good |
| Frequency | Rare |
Angiofibroma of soft tissue (AFST), also termed angiofibroma, not otherwise specified, is a rare disorder that was classified in the category of benign fibroblastic and myofibroblastic tumors by the World Health Organization in 2020.[1] AFST tumors typically occur in a leg but can occur in other locations; they develop in older children and adults including elderly individuals. AFSTs are slow-growing, often painless tumors composed primarily of spindle-shaped cells and a prominent vascular network. The spindle-shaped cells are benign tumor cells that in almost all cases have chromosome abnormalities that are thought to contribute to their abnormal development and/or growth.[2]
AFST tumors are commonly treated by surgical excision although in uncommon cases they recur at the site of their removal and require further surgical treatment.[3] They do not metastasize to distant tissues and overall have a good prognosis.[4]
An AFST tumor is a neoplasm (i.e. growth of tissue that is not coordinated with the normal surrounding tissue and persists in growing even if the original trigger for growth is removed) that was first described by A. Mariño-Enríquez and C.D. Fletcher in 2012.[3]
Signs and symptoms
AFST tumors commonly present as slowly growing, painless, deep-seated lumps in individuals aged 6 to 86 years old (median age 47-50 years in different studies).[4] The tumors are most common in the lower extremities but uncommonly occur in the back, chest wall, iliac crest, groin and nearby lower lateral abdominal region,[4] abdominal cavity, pelvic cavity,[5] breast,[2] cheek, temporal region of the head and, in a report on 24 AFST cases done in Shanghai, the upper limb in 3 cases and, in 1 case each, the retroperitoneum and liver.[6] The tumors' longest diameters have ranged from 1.2 to 10 cm (mean: 5.1 cm)[4] and 0.8 to 14 cm (mean: 4.6 cm)[6] in two different studies.
Pathology
Grossly, AFST tumors, when visible on skin, are located in the skin's subcutaneous tissue or the fascia layer below the subcutaneous tissue.[2] They may be infiltrating deeper into these tissues[4] and/or into nearby large joints.[2] Regardless of location, however, most of these tumors are well-circumscribed.[5]
Histopathological microscopic analyses of hematoxylin and eosin-stained AFST tissues generally reveal bland appearing spindle-shaped cells and a prominent small, thin-walled blood vessels network in a background of alternating myxoid connective tissue areas and more highly cellular collagen fiber-rich connective tissue areas. (Myxoid indicates areas that appear more blue or purple than normal due to their high uptake of the hematoxylin stain.) Typically, these tumors appear well-circumscribed but some cases show them infiltrating into adjacent normal adipose tissues, connective tissues, skeletal muscles, and/or joints.[4] Immunohistochemistry analyses (i.e. identifying specific proteins in cells using antibodies that bind to these proteins) of AFST tissues detect cells bearing estrogen receptor, CD163 and NCOA2 proteins in 100% of cases; MUC1 (also termed EMA) protein in 46% of cases; desmin protein in 22% of cases; ACTA2 (also termed α-SMA), CD34, and STAT6 proteins in 10% or fewer cases, and S100 and cytokeratin proteins in no cases.
Chromosome and gene abnormalities
In 60-80% of cases, the cells in AFST tumors express the AHRR-NCOA2 fusion gene. A fusion gene is a hybrid gene formed from two previously independent genes as a result of a translocation, interstitial deletion, or chromosomal inversion.[5] The AHRR gene (i.e. gene for the aryl hydrocarbon receptor repressor protein) is located at band 15.33 on the short (or "p") arm of chromosome 5 (cite designation: 5p15.33);[7] The NCOA2 gene (i.e. gene for the nuclear receptor coactivator 2 protein) is located at band 13.3 on the long (or "q") arm of chromosome 8 (cite designation: 8q13.3).[8] A translocation between these two chromosomes creates the AHRR-NCOA2 fusion gene (fusion gene designation: t(5;8)(q15;q13)).[9] AHRR is a tumor suppressor gene that when fused to other genes is found in the cells of , and thought to promote, various leukemias and neoplasms. It is thought to similarly promote the development and/or progression of AFST tumors.[4] While most commonly associated with the AHRR-NCOA2 fusion gene, rare AFST tumor cases have also been shown to be associated with GAB1-ABL1, GTF2I-NCOA2, NCOA2-ETV4, ETV4-AHRR,[4] and NAB2-STAT6[4] fusion genes.
Diagnosis
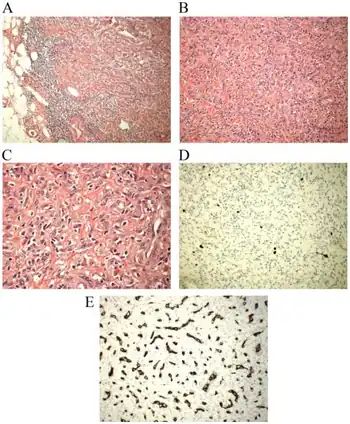
The diagnosis of AFST depends on its presentation (particularly its location), histopathology (particularly the expression of certain proteins by its tumor cells), and the presence of specific fusion genes (e.g. AGRR-NCOA2) in its tumor cells. Among benign tumors, cellular angiofibroma and solitary fibrous tumor may be confused with AFST. Cellular angiofibroma differs from AFTS in its typical location (i.e. inguinal, scrotal, and vulva areas), its distinct histology of rounded, non-branching vessels, high cellularity, and cells with small nuclei, and its tumor cells' loss of the RB1 gene. Solitary fibrous tumors differ from AFST tumors in their common location in the lungs' pleurae, their characteristic branching dilated, staghorn-shaped blood vessels, and their tumor cells' expression of CD34 and STAT6 proteins in the majority of the cases. AFTS tumors may also be confused with three malignant tumors, low-grade fibromyxoid sarcoma, myxofibrosarcoma, and myxoid liposarcoma. Low-grade fibromyxoid sarcomas tend to be less cellular and have less prominent blood vessel than AFST; they also differ from AFST in that their tumor cells commonly express the MUC4 protein and FUS-CREB3L1, FUS-CREBL2, or, EWSR1-CREB3L1 fusion genes. Myxofibrosarcoma tumors commonly show overt malignant features such as highly infiltrating margins, tumor cells with eosinophilic cytoplasm, atypical nuclei, and rapid proliferation rates as evidenced by their high mitotic indexes. Myxoid liposarcoma tumors consist of round or slightly fusiform cells in a myxoid matrix, vacuolated lipoblasts (i.e. cells that are precursors to fat cells), and arborizing networks of thin-walled capillaries.[2] In all of these cases, the presence of one of the AFST-associated fusion genes cited in the previous section lends support for the diagnosis of AFST.[2][9]
Treatment
AFST tumors are typically treated by total surgical resection in order to remove all tumor tissue.[4] Uncommonly, these tumors have recurred at the site of their removal, particularly in cases where a portion of the original tumor was not removed.[5] Recurrences have occurred 4-120 months after the original resections[4] and have been treated by a second surgical resection.[3]
Prognosis
Overall, AFST tumors have a good prognosis.[4]
References
- ↑ Sbaraglia M, Bellan E, Dei Tos AP (April 2021). "The 2020 WHO Classification of Soft Tissue Tumours: news and perspectives". Pathologica. 113 (2): 70–84. doi:10.32074/1591-951X-213. PMC 8167394. PMID 33179614.
- 1 2 3 4 5 6 Kallen ME, Hornick JL (January 2021). "The 2020 WHO Classification: What's New in Soft Tissue Tumor Pathology?". The American Journal of Surgical Pathology. 45 (1): e1–e23. doi:10.1097/PAS.0000000000001552. PMID 32796172. S2CID 225430576.
- 1 2 3 Mariño-Enríquez A, Fletcher CD (April 2012). "Angiofibroma of soft tissue: clinicopathologic characterization of a distinctive benign fibrovascular neoplasm in a series of 37 cases". The American Journal of Surgical Pathology. 36 (4): 500–8. doi:10.1097/PAS.0b013e31823defbe. PMID 22301504. S2CID 45742809.
- 1 2 3 4 5 6 7 8 9 10 11 12 Mindiola-Romero AE, Maloney N, Bridge JA, Korkolopoulou P, Sakellariou S, Linos K (February 2020). "A concise review of angiofibroma of soft tissue: A rare newly described entity that can be encountered by dermatopathologists". Journal of Cutaneous Pathology. 47 (2): 179–185. doi:10.1111/cup.13580. PMID 31568567. S2CID 203625513.
- 1 2 3 4 Ali Z, Anwar F (November 2019). "Angiofibroma of Soft Tissue: A Newly Described Entity; A Case Report and Review of Literature". Cureus. 11 (11): e6225. doi:10.7759/cureus.6225. PMC 6929243. PMID 31890425.
- 1 2 Xu XL, Liu JG, Sun M, Yu L, Liu QY, Bai QM, Wu LJ, Wang J (August 2018). "[Angiofibroma of soft tissue: a clinicopathologic analysis of 24 cases]". Zhonghua Bing Li Xue Za Zhi = Chinese Journal of Pathology (in Chinese). 47 (8): 616–621. doi:10.3760/cma.j.issn.0529-5807.2018.08.009. PMID 30107667.
{{cite journal}}: CS1 maint: unrecognized language (link) - ↑ "AHRR aryl-hydrocarbon receptor repressor [Homo sapiens (Human)] - Gene - NCBI". Archived from the original on 2021-10-31. Retrieved 2022-02-19.
- ↑ "NCOA2 nuclear receptor coactivator 2 [Homo sapiens (Human)] - Gene - NCBI". Archived from the original on 2022-01-19. Retrieved 2022-02-19.
- 1 2 Oda Y, Yamamoto H, Kohashi K, Yamada Y, Iura K, Ishii T, Maekawa A, Bekki H (September 2017). "Soft tissue sarcomas: From a morphological to a molecular biological approach". Pathology International. 67 (9): 435–446. doi:10.1111/pin.12565. PMID 28759137. S2CID 34316562.