Beare–Stevenson cutis gyrata syndrome
| Beare–Stevenson cutis gyrata syndrome | |
|---|---|
| Other names: Cutis gyrata-acanthosis nigricans-craniosynostosis syndrome[1] | |
| Diagnostic method | identification of the p.Pro250Arg pathogenic variant in FGFR3; the diagnosis of FGFR2-related isolated coronal synostosis is based on identification of a FGFR2 pathogenic variant. The diagnosis of the other six FGFR-related craniosynostosis syndromes is based on clinical findings; molecular genetic testing of FGFR1, FGFR2, and FGFR3 may be helpful in establishing the specific diagnosis in questionable cases. |
Beare–Stevenson cutis gyrata syndrome is a rare genetic disorder characterized by craniosynostosis (premature fusion of certain bones of the skull, sometimes resulting in a characteristic 'cloverleaf skull'; further growth of the skull is prevented, and therefore the shape of the head and face is abnormal) and a specific skin abnormality, called cutis gyrata, characterized by a furrowed and wrinkled appearance (particularly in the face and on the palms and soles of the feet); thick, dark, velvety areas of skin (acanthosis nigricans) are sometimes found on the hands and feet and in the groin.[2][3]
Signs and symptoms
Signs and symptoms of Beare–Stevenson cutis gyrata syndrome can include a blockage of the nasal passages (choanal atresia), overgrowth of the umbilical stump, and abnormalities of the genitalia and anus. The medical complications associated with this condition are often severe and may well be life-threatening in infancy or early childhood.
Genetics
Several mutations in the FGFR2 gene (a gene coding for a protein called fibroblast growth factor receptor 2, which is involved in important signaling pathways) are known to cause Beare–Stevenson cutis gyrata syndrome;[3] however, not all patients with the condition have a mutation in their FGFR2 gene. Any alternative underlying causes are currently unidentified. The syndrome follows an autosomal dominant pattern, meaning that if one of the two available genes carries a mutation the syndrome will result. Currently, no familial histories are known (in other words, there are no reports of cases in which a parent carrying a mutation in their FGFR2 gene then propagated said mutation to his or her child).
Diagnosis
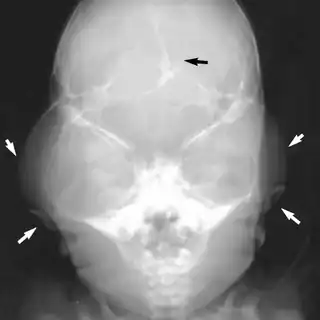
The evaluation of Bears Stevenson cutis gyrata syndrome is done via:[4]
- Medical exam
- Clinical presentation
- Medical history
- Lab exam
- Genetic test
Prognosis
The prognosis for this condition is poor as medical complications are often life-threatening very early in life[5]
Incidence
Beare–Stevenson cutis gyrata syndrome is so rare that a reliable incidence cannot be established as of yet; fewer than 25 patients with the condition have been reported.
See also
References
- ↑ RESERVED, INSERM US14-- ALL RIGHTS. "Orphanet: Cutis gyrata acanthosis nigricans craniosynostosis syndrome". www.orpha.net. Archived from the original on 29 July 2019. Retrieved 14 March 2019.
- ↑ http://ghr.nlm.nih.gov/condition/beare-stevenson-cutis-gyrata-syndrome Archived 2020-09-28 at the Wayback Machine The Genetic Home Reference entry on Beare-Stevenson cutis gyrata syndrome
- 1 2 Hall BD, Cadle RG, Golabi M, Morris CA, Cohen MM (September 1992). "Beare-Stevenson cutis gyrata syndrome". Am J Med Genet. 44 (1): 82–89. doi:10.1002/ajmg.1320440120. PMID 1519658.
- ↑ "Beare-Stevenson cutis gyrata syndrome | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Archived from the original on 26 November 2021. Retrieved 23 November 2021.
- ↑ "Beare-Stevenson cutis gyrata syndrome: MedlinePlus Genetics". medlineplus.gov. Archived from the original on 16 April 2021. Retrieved 23 November 2021.
External links
| Classification | |
|---|---|
| External resources |
|