Biochemistry of Alzheimer's disease
The biochemistry of Alzheimer's disease, the most common cause of dementia, is not yet very well understood. Alzheimer's disease (AD) has been identified as a proteopathy: a protein misfolding disease due to the accumulation of abnormally folded amyloid beta (Aβ) protein in the brain.[1] Amyloid beta is a short peptide that is an abnormal proteolytic byproduct of the transmembrane protein amyloid-beta precursor protein (APP), whose function is unclear but thought to be involved in neuronal development.[2] The presenilins are components of proteolytic complex involved in APP processing and degradation.[3][4]
Amyloid beta monomers are soluble and contain short regions of beta sheet and polyproline II helix secondary structures in solution,[5] though they are largely alpha helical in membranes;[6] however, at sufficiently high concentration, they undergo a dramatic conformational change to form a beta sheet-rich tertiary structure that aggregates to form amyloid fibrils.[7] These fibrils and oligomeric forms of Aβ deposit outside neurons in formations known as senile plaques. There are different types of plaques, including the diffuse, compact, cored or neuritic plaque types, as well as Aβ deposits in the walls of small blood vessel walls in the brain called cerebral amyloid angiopathy.[8] [9]
AD is also considered a tauopathy due to abnormal aggregation of the tau protein, a microtubule-associated protein expressed in neurons that normally acts to stabilize microtubules in the cell cytoskeleton. Like most microtubule-associated proteins, tau is normally regulated by phosphorylation; however, in Alzheimer's disease, hyperphosphorylated tau accumulates as paired helical filaments[10] that in turn aggregate into masses inside nerve cell bodies known as neurofibrillary tangles and as dystrophic neurites associated with amyloid plaques. Although little is known about the process of filament assembly, depletion of a prolyl isomerase protein in the parvulin family has been shown to accelerate the accumulation of abnormal tau.[11][12]
Neuroinflammation is also involved in the complex cascade leading to AD pathology and symptoms. Considerable pathological and clinical evidence documents immunological changes associated with AD, including increased pro-inflammatory cytokine concentrations in the blood and cerebrospinal fluid.[13][14] Whether these changes may be a cause or consequence of AD remains to be fully understood, but inflammation within the brain, including increased reactivity of the resident microglia towards amyloid deposits, has been implicated in the pathogenesis and progression of AD.[15]
Neuropathology
At a macroscopic level, AD is characterized by loss of neurons and synapses in the cerebral cortex and certain subcortical regions. This results in gross atrophy of the affected regions, including degeneration in the temporal lobe and parietal lobe, and parts of the frontal cortex and cingulate gyrus.[16]
Both amyloid plaques and neurofibrillary tangles are clearly visible by microscopy in AD brains.[17] Plaques are dense, mostly insoluble deposits of protein and cellular material outside and around neurons. Tangles are insoluble twisted fibers that build up inside the nerve cell. Though many older people develop some plaques and tangles, the brains of AD patients have them to a much greater extent and in different brain locations.[18]
Biochemical characteristics
Fundamental to the understanding of Alzheimer's disease is the biochemical events that leads to accumulation of the amyloid-beta and tau-protein. A delicate balance of the enzymes secretases regulate the amyloid-beta accumulation. Alpha Secretase can render a non-pathological (non-amyloidogenic) Amyloid Beta (DOI: 10.2174/156720512799361655). Recently, a link between cholinergic neuronal activity and the activity of alpha-secretase has been highlighted,[19] which can discourage Amyloid-beta proteins deposition in brain of patients with Alzheimer's disease. Alzheimer's disease has been identified as a protein misfolding disease, or proteopathy, due to the accumulation of abnormally folded Amyloid-beta proteins in the brains of AD patients.[1] Abnormal amyloid-beta accumulation can first be detected using cerebrospinal fluid analysis and later using positron emission tomography (PET).[20]
Although AD shares pathophysiological mechanisms with prion diseases, it is not transmissible like prion diseases.[21] Amyloid-beta, also written Aβ, is a short peptide that is a proteolytic byproduct of the transmembrane protein amyloid precursor protein (APP), whose function is unclear but thought to be involved in neuronal development. The presenilins are components of a proteolytic complex involved in APP processing and degradation.[4] Although amyloid beta monomers are harmless, they undergo a dramatic conformational change at sufficiently high concentration to form a beta sheet-rich tertiary structure that aggregates to form amyloid fibrils[7] that deposit outside neurons in dense formations known as senile plaques or neuritic plaques, in less dense aggregates as diffuse plaques, and sometimes in the walls of small blood vessels in the brain in a process called amyloid angiopathy or congophilic angiopathy.
AD is also considered a tauopathy due to abnormal aggregation of the tau protein, a microtubule-associated protein expressed in neurons that normally acts to stabilize microtubules in the cell cytoskeleton. Like most microtubule-associated proteins, tau is normally regulated by phosphorylation; however, in AD patients, hyperphosphorylated tau accumulates as paired helical filaments[10] that in turn aggregate into masses inside nerve cell bodies known as neurofibrillary tangles and as dystrophic neurites associated with amyloid plaques.
Levels of the neurotransmitter acetylcholine (ACh) are reduced. Levels of other neurotransmitters serotonin, norepinephrine, and somatostatin are also often reduced. Replenishing the ACh by anti-cholinesterases is an approved mode of treatment by FDA. An alternative method of stimulating ACh receptors of M1-M3 types by synthetic agonists that have a slower rate of dissociation from the receptor has been proposed as next generation cholinomimetic in Alzheimer's disease[15].
Potential disease mechanisms
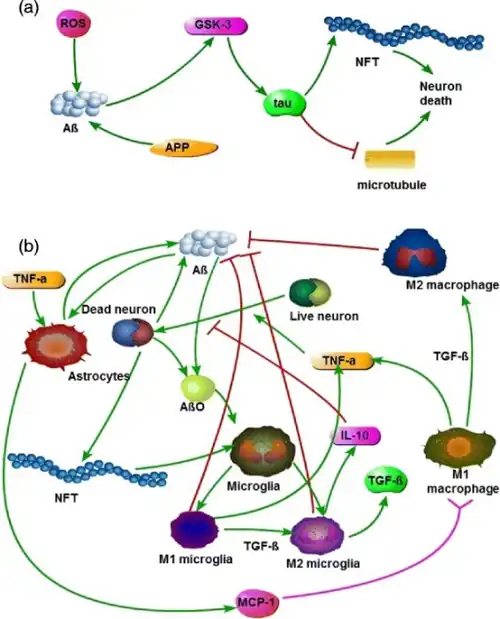
While the gross histological features of AD in the brain have been well characterized, several different hypotheses have been advanced regarding the primary cause. Among the oldest hypotheses is the cholinergic hypothesis, which suggests that deficiency in cholinergic signaling initiates the progression of the disease.[22] Other hypotheses suggest that either misfolding tau protein inside the cell or aggregation of amyloid beta outside the cell initiates the cascade leading to AD pathology[23][24]>. Still other hypotheses propose metabolic factors,[25] vascular disturbance,[26] or chronically elevated inflammation in the brain[27] as the primary cause for AD. While researchers have not identified a clear causative pathway originating from any of the molecular hypothesis that explains the gross anatomical changes observed in advanced AD, variants of the amyloid beta hypothesis of molecular initiation have become dominant among many researchers to date[28]
Cholinergic hypothesis
The cholinergic hypothesis of AD development was first proposed in 1976 by Peter Davies and A.J.F Maloney.[29] It states that Alzheimer's begins as a deficiency in the production of acetylcholine, a vital neurotransmitter. Much early therapeutic research was based on this hypothesis, including restoration of the "cholinergic nuclei". The possibility of cell-replacement therapy was investigated on the basis of this hypothesis. All of the first-generation anti-Alzheimer's medications are based on this hypothesis and work to preserve acetylcholine by inhibiting acetylcholinesterases (enzymes that break down acetylcholine). These medications, though sometimes beneficial, have not led to a cure. In all cases, they have served to only treat symptoms of the disease and have neither halted nor reversed it. These results and other research have led to the conclusion that acetylcholine deficiencies may not be directly causal, but are a result of widespread brain tissue damage, damage so widespread that cell-replacement therapies are likely to be impractical.
More recent hypotheses center on the effects of the misfolded and aggregated proteins, amyloid beta and tau. The two positions are lightheartedly described as "ba-ptist" and "tau-ist" viewpoints in one scientific publication. Therein, it is suggested that "Tau-ists" believe that the tau protein abnormalities initiate the disease cascade, while "ba-ptists" believe that beta amyloid deposits are the causative factor in the disease.[30]
Tau hypothesis
The hypothesis that tau is the primary causative factor has long been grounded in the observation that deposition of amyloid plaques does not correlate well with neuron loss.[31] A mechanism for neurotoxicity has been proposed based on the loss of microtubule-stabilizing tau protein that leads to the degradation of the cytoskeleton.[32] However, consensus has not been reached on whether tau hyperphosphorylation precedes or is caused by the formation of the abnormal helical filament aggregates.[30] Support for the tau hypothesis also derives from the existence of other diseases known as tauopathies in which the same protein is identifiably misfolded.[33] However, a majority of researchers support the alternative hypothesis that amyloid is the primary causative agent.[30]
Amyloid hypothesis
The amyloid hypothesis is initially compelling because the gene for the amyloid beta precursor APP is located on chromosome 21, and patients with trisomy 21 - better known as Down syndrome - who have an extra gene copy exhibit AD-like disorders by 40 years of age.[34][35] The amyloid hypothesis points to the cytotoxicity of mature aggregated amyloid fibrils, which are believed to be the toxic form of the protein responsible for disrupting the cell's calcium ion homeostasis and thus inducing apoptosis.[36] This hypothesis is supported by the observation that higher levels of a variant of the beta amyloid protein known to form fibrils faster in vitro correlate with earlier onset and greater cognitive impairment in mouse models[37] and with AD diagnosis in humans.[38] However, mechanisms for the induced calcium influx, or proposals for alternative cytotoxic mechanisms, by mature fibrils are not obvious.
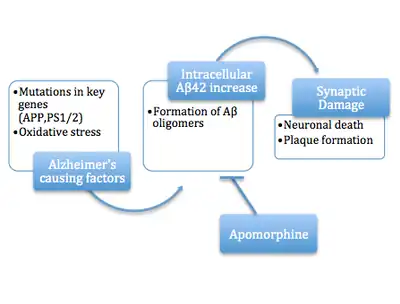
A more recent variation of the amyloid hypothesis identifies the cytotoxic species as an intermediate misfolded form of amyloid beta, neither a soluble monomer nor a mature aggregated polymer but an oligomeric species, possibly toroidal or star-shaped with a central channel[39] that may induce apoptosis by physically piercing the cell membrane.[40] This ion channel hypothesis postulates that oligomers of soluble, non-fibrillar Aβ form membrane ion channels allowing unregulated calcium influx into neurons.[41] A related alternative suggests that a globular oligomer localized to dendritic processes and axons in neurons is the cytotoxic species.[42][43] The prefibrillar aggregates were shown to be able to disrupt the membrane.[44]
The cytotoxic-fibril hypothesis presents a clear target for drug development: inhibit the fibrillization process. Much early development work on lead compounds has focused on this inhibition;[45][46][47] most are also reported to reduce neurotoxicity, but the toxic-oligomer theory would imply that prevention of oligomeric assembly is the more important process[48][49] [50] or that a better target lies upstream, for example in the inhibition of APP processing to amyloid beta.[51] For example, apomorphine was seen to significantly improve memory function through the increased successful completion of the Morris Water Maze.[48]
- Soluble intracellular (o)Aβ42
Two papers have shown that oligomeric (o)Aβ42 (a species of Aβ), in soluble intracellular form, acutely inhibits synaptic transmission, a pathophysiology that characterizes AD (in its early stages), by activating casein kinase 2.[52][53]
Inflammatory Hypothesis
Converging evidence suggests/supports that a sustained inflammatory response in the brain is a core feature of AD pathology and may be a key factor in AD pathogenesis.[54][55] The brains of AD patients exhibit several markers of increased inflammatory signaling.[56][57][58] The inflammatory hypothesis proposes that chronically elevated inflammation in the brain is a crucial component to the amyloid cascade[59] in the early phases of AD and magnifies disease severity in later stages of AD. Aβ is present in healthy brains and serves a vital physiological function in recovery from neuronal injury, protection from infection, and repair of the blood-brain barrier,[60] however it is unknown how Aβ production starts to exceed the clearance capacity of the brain and initiates AD progression. A possible explanation is that Aβ causes microglia, the resident immune cell of the brain, to become activated and secrete pro-inflammatory signaling molecules, called cytokines, which recruit other local microglia.[61] While acute microglial activation, as in response to injury, is beneficial and allows microglia to clear Aβ and other cellular debris via phagocytosis, chronically activated microglia exhibit decreased efficiency in Aβ clearance.[62] Despite this reduced AB clearance capacity, activated microglia continue to secrete pro-inflammatory cytokines like interleukins 1β and 6 (IL-6, IL-1β) and tumor necrosis factor-alpha (TNF-a), as well as reactive oxygen species which disrupt healthy synaptic functioning[63] and eventually cause neuronal death.[64] The loss of synaptic functioning and later neuronal death is responsible for the cognitive impairments and loss of volume in key brain regions which are associated with AD.[65] IL-1B, IL-6, and TNF-a cause further production of Aβ oligomers, as well as tau hyperphosphorylation, leading to continued microglia activation and creating a feed forward mechanism in which Aβ production is increased and Aβ clearance is decreased eventually causing the formation of Aβ plaques.[66][67]
Isoprenoid changes
A 1994 study [68] showed that the isoprenoid changes in Alzheimer's disease differ from those occurring during normal aging and that this disease cannot, therefore, be regarded as a result of premature aging. During aging the human brain shows a progressive increase in levels of dolichol, a reduction in levels of ubiquinone, but relatively unchanged concentrations of cholesterol and dolichyl phosphate. In Alzheimer's disease, the situation is reversed with decreased levels of dolichol and increased levels of ubiquinone. The concentrations of dolichyl phosphate are also increased, while cholesterol remains unchanged. The increase in the sugar carrier dolichyl phosphate may reflect an increased rate of glycosylation in the diseased brain and the increase in the endogenous anti-oxidant ubiquinone an attempt to protect the brain from oxidative stress, for instance induced by lipid peroxidation.[69] Ropren, identified previously in Russia, is neuroprotective in a rat model of Alzheimer's disease.[70][71]
Glucose consumption
The human brain is one of the most metabolically active organs in the body and metabolizes a large amount of glucose to produce cellular energy in the form of adenosine triphosphate (ATP).[72] Despite its high energy demands, the brain is relatively inflexible in its ability to utilize substrates for energy production and relies almost entirely on circulating glucose for its energy needs.[73] This dependence on glucose puts the brain at risk if the supply of glucose is interrupted, or if its ability to metabolize glucose becomes defective. If the brain is not able to produce ATP, synapses cannot be maintained and cells cannot function, ultimately leading to impaired cognition.[73]
Imaging studies have shown decreased utilization of glucose in the brains of Alzheimer’s disease patients early in the disease, before clinical signs of cognitive impairment occur. This decrease in glucose metabolism worsens as clinical symptoms develop and the disease progresses.[74][75] Studies have found a 17%-24% decline in cerebral glucose metabolism in patients with Alzheimer’s disease, compared with age-matched controls.[76] Numerous imaging studies have since confirmed this observation.
Abnormally low rates of cerebral glucose metabolism are found in a characteristic pattern in the Alzheimer’s disease brain, particularly in the posterior cingulate, parietal, temporal, and prefrontal cortices. These brain regions are believed to control multiple aspects of memory and cognition. This metabolic pattern is reproducible and has even been proposed as a diagnostic tool for Alzheimer’s disease. Moreover, diminished cerebral glucose metabolism (DCGM) correlates with plaque density and cognitive deficits in patients with more advanced disease.[76][77]
Diminished cerebral glucose metabolism (DCGM) may not be solely an artifact of brain cell loss since it occurs in asymptomatic patients at risk for Alzheimer’s disease, such as patients homozygous for the epsilon 4 variant of the apolipoprotein E gene (APOE4, a genetic risk factor for Alzheimer’s disease), as well as in inherited forms of Alzheimer’s disease.[78] Given that DCGM occurs before other clinical and pathological changes occur, it is unlikely to be due to the gross cell loss observed in Alzheimer’s disease.[73]
In imaging studies involving young adult APOE4 carriers, where there were no signs of cognitive impairment, diminished cerebral glucose metabolism (DCGM) was detected in the same areas of the brain as older subjects with Alzheimer’s disease.[78] However, DCGM is not exclusive to APOE4 carriers. By the time Alzheimer’s has been diagnosed, DCGM occurs in genotypes APOE3/E4, APOE3/E3, and APOE4/E4.[79] Thus, DCGM is a metabolic biomarker for the disease state.[80]
Insulin signaling
A connection has been established between Alzheimer's disease and diabetes during the past decade, as insulin resistance, which is a characteristic hallmark of diabetes, has also been observed in brains of subjects suffering from Alzheimer's disease.[81] Neurotoxic oligomeric amyloid-β species decrease the expression of insulin receptors on the neuronal cell surface[82] and abolish neuronal insulin signaling.[81] It has been suggested that neuronal gangliosides, which take part in the formation of membrane lipid microdomains, facilitate amyloid-β-induced removal of the insulin receptors from the neuronal surface.[83] In Alzheimer's disease, oligomeric amyloid-β species trigger TNF-α signaling.[81] c-Jun N-terminal kinase activation by TNF-α in turn activates stress-related kinases and results in IRS-1 serine phosphorylation, which subsequently blocks downstream insulin signaling.[81][84][85] The resulting insulin resistance contributes to cognitive impairment. Consequently, increasing neuronal insulin sensitivity and signaling may constitute a novel therapeutic approach to treat Alzheimer's disease.[86][87]
Oxidative stress
Oxidative stress is emerging as a key factor in the pathogenesis of AD.[88] Reactive oxygen species (ROS) over-production is thought to play a critical role in the accumulation and deposition of amyloid beta in AD.[89] Brains of AD patients have elevated levels of oxidative DNA damage in both nuclear and mitochondrial DNA, but the mitochondrial DNA has approximately 10-fold higher levels than nuclear DNA.[90] Aged mitochondria may be the critical factor in the origin of neurodegeneration in AD.[89] Even individuals with mild cognitive impairment, the phase between normal aging and early dementia, have increased oxidative damage in their nuclear and mitochondrial brain DNA[91] (see Aging brain).
Large gene instability hypothesis
A bioinformatics analysis in 2017[92] revealed that extremely large human genes are significantly over-expressed in brain and take part in the postsynaptic architecture. These genes are also highly enriched in cell adhesion Gene Ontology (GO) terms and often map to chromosomal fragile sites.[93] The majority of known Alzheimer's disease risk gene products including the amyloid precursor protein (APP) and gamma-secretase, as well as the APOE receptors and GWAS risk loci take part in similar cell adhesion mechanisms. It was concluded that dysfunction of cell and synaptic adhesion is central to Alzheimer's disease pathogenesis, and mutational instability of large synaptic adhesion genes may be the etiological trigger of neurotransmission disruption and synaptic loss in brain aging. As a typical example, this hypothesis explains the APOE risk locus of AD in context of signaling of its giant lipoprotein receptor, LRP1b which is a large tumor-suppressor gene with brain-specific expression and also maps to an unstable chromosomal fragile site. The large gene instability hypothesis puts the DNA damage mechanism at the center of Alzheimer's disease pathophysiology.
DNA damage
Naturally occurring DNA double-strand breaks (DSBs) arise in human cells largely from single-strand breaks induced by various processes including the activity of reactive oxygen species, topoisomerases, and hydrolysis due to thermal fluctuations.[94] In neurons DSBs are induced by a type II topoisomerase as part of the physiologic process of memory formation.[95] DSBs are present in both neurons and astrocytes in the postmortem human hippocampus of AD patients at a higher level than in non-AD individuals.[96] AD is associated with an accumulation of DSBs in neurons and astrocytes in the hippocampus and frontal cortex from early stages onward.[97] DSBs are increased in the vicinity of amyloid plaques in the hippocampus, indicating a potential role for Aβ in DSB accumulation or vice versa.[96] The predominant mechanism for repairing DNA double-strand breaks is non-homologous end joining (NHEJ), a mechanism that utilizes the DNA-dependent protein kinase (DNA-PK) complex. The end joining activity and protein levels of DNA-PK catalytic subunit are significantly lower in AD brains than in normal brains.[98]
References
- 1 2 Hashimoto M, Rockenstein E, Crews L, Masliah E (2003). "Role of protein aggregation in mitochondrial dysfunction and neurodegeneration in Alzheimer's and Parkinson's diseases". Neuromolecular Medicine. 4 (1–2): 21–36. doi:10.1385/NMM:4:1-2:21. PMID 14528050. S2CID 20760249.
- ↑ Kerr ML, Small DH (April 2005). "Cytoplasmic domain of the beta-amyloid protein precursor of Alzheimer's disease: function, regulation of proteolysis, and implications for drug development". Journal of Neuroscience Research. 80 (2): 151–9. doi:10.1002/jnr.20408. PMID 15672415. S2CID 31985212.
- ↑ Borchelt DR (January 1998). "Metabolism of presenilin 1: influence of presenilin 1 on amyloid precursor protein processing". Neurobiology of Aging. 19 (1 Suppl): S15-8. doi:10.1016/S0197-4580(98)00026-8. PMID 9562461. S2CID 4000041.
- 1 2 Cai D, Netzer WJ, Zhong M, Lin Y, Du G, Frohman M, et al. (February 2006). "Presenilin-1 uses phospholipase D1 as a negative regulator of beta-amyloid formation". Proceedings of the National Academy of Sciences of the United States of America. 103 (6): 1941–6. Bibcode:2006PNAS..103.1941C. doi:10.1073/pnas.0510708103. PMC 1413665. PMID 16449386.
- ↑ Danielsson J, Andersson A, Jarvet J, Gräslund A (July 2006). "15N relaxation study of the amyloid beta-peptide: structural propensities and persistence length". Magnetic Resonance in Chemistry. 44 Spec No: S114-21. doi:10.1002/mrc.1814. PMID 16826550. S2CID 26462689.
- ↑ Tomaselli S, Esposito V, Vangone P, van Nuland NA, Bonvin AM, Guerrini R, et al. (February 2006). "The alpha-to-beta conformational transition of Alzheimer's Abeta-(1-42) peptide in aqueous media is reversible: a step by step conformational analysis suggests the location of beta conformation seeding". ChemBioChem. 7 (2): 257–67. doi:10.1002/cbic.200500223. hdl:1874/20092. PMID 16444756. S2CID 84875550.
- 1 2 Ohnishi S, Takano K (March 2004). "Amyloid fibrils from the viewpoint of protein folding". Cellular and Molecular Life Sciences. 61 (5): 511–524. doi:10.1007/s00018-003-3264-8. PMID 15004691. S2CID 25739126.
- ↑ Duyckaerts, Charles; Dickson, Dennis W. (2011). Neurodegeneration: the molecular pathology of dementia and movement disorders. Oxford: Wiley-Blackwell. p. 62-91.
- ↑ Röhr D, Boon BD (December 2020). "Label-free vibrational imaging of different Aβ plaque types in Alzheimer's disease reveals sequential events in plaque development". Acta Neuropathologica Communications. 8 (1): 222. doi:10.1186/s40478-020-01091-5. PMC 7733282. PMID 33308303.
- 1 2 Goedert M, Klug A, Crowther RA (2006). "Tau protein, the paired helical filament and Alzheimer's disease". Journal of Alzheimer's Disease. 9 (3 Suppl): 195–207. doi:10.3233/JAD-2006-9S323. PMID 16914859.
- ↑ Pastorino L, Sun A, Lu PJ, Zhou XZ, Balastik M, Finn G, et al. (March 2006). "The prolyl isomerase Pin1 regulates amyloid precursor protein processing and amyloid-beta production". Nature. 440 (7083): 528–34. Bibcode:2006Natur.440..528P. doi:10.1038/nature04543. PMID 16554819. S2CID 4421584.
- ↑ Lim J, Lu KP (January 2005). "Pinning down phosphorylated tau and tauopathies". Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 1739 (2–3): 311–22. doi:10.1016/j.bbadis.2004.10.003. PMID 15615648.
- ↑ Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, et al. (2000). "Inflammation and Alzheimer's disease". Neurobiology of Aging. 21 (3): 383–421. doi:10.1016/S0197-4580(00)00124-X. PMC 3887148. PMID 10858586.
- ↑ Swardfager W, Lanctôt K, Rothenburg L, Wong A, Cappell J, Herrmann N (November 2010). "A meta-analysis of cytokines in Alzheimer's disease". Biological Psychiatry. 68 (10): 930–41. doi:10.1016/j.biopsych.2010.06.012. PMID 20692646. S2CID 6544784.
- ↑ Vasefi M, Hudson M, Ghaboolian-Zare E (November 2019). "Diet Associated with Inflammation and Alzheimer's Disease". Journal of Alzheimer's Disease Reports. 3 (1): 299–309. doi:10.3233/ADR-190152. PMC 6918878. PMID 31867568.
- ↑ Wenk GL (2003). "Neuropathologic changes in Alzheimer's disease". The Journal of Clinical Psychiatry. 64 Suppl 9: 7–10. PMID 12934968.
- ↑ Tiraboschi P, Hansen LA, Thal LJ, Corey-Bloom J (June 2004). "The importance of neuritic plaques and tangles to the development and evolution of AD". Neurology. 62 (11): 1984–9. doi:10.1212/01.WNL.0000129697.01779.0A. PMID 15184601. S2CID 25017332.
- ↑ Bouras C, Hof PR, Giannakopoulos P, Michel JP, Morrison JH (1994). "Regional distribution of neurofibrillary tangles and senile plaques in the cerebral cortex of elderly patients: a quantitative evaluation of a one-year autopsy population from a geriatric hospital". Cerebral Cortex. 4 (2): 138–50. doi:10.1093/cercor/4.2.138. PMID 8038565.
- ↑ Baig AM (January 2019). "Connecting the Dots: Linking the Biochemical to Morphological Transitions in Alzheimer's Disease". ACS Chemical Neuroscience. 10 (1): 21–24. doi:10.1021/acschemneuro.8b00409. PMID 30160095.
- ↑ Palmqvist S, Mattsson N, Hansson O (April 2016). "Cerebrospinal fluid analysis detects cerebral amyloid-β accumulation earlier than positron emission tomography". Brain. 139 (Pt 4): 1226–36. doi:10.1093/brain/aww015. PMC 4806222. PMID 26936941.
- ↑ Castellani RJ, Perry G, Smith MA (2004). "Prion disease and Alzheimer's disease: pathogenic overlap". Acta Neurobiologiae Experimentalis. 64 (1): 11–7. PMID 15190676.
- ↑ Francis PT, Palmer AM, Snape M, Wilcock GK (February 1999). "The cholinergic hypothesis of Alzheimer's disease: a review of progress". Journal of Neurology, Neurosurgery, and Psychiatry. 66 (2): 137–47. doi:10.1136/jnnp.66.2.137. PMC 1736202. PMID 10071091.
- ↑ Tanzi RE, Bertram L (February 2005). "Twenty years of the Alzheimer's disease amyloid hypothesis: a genetic perspective". Cell. 120 (4): 545–55. doi:10.1016/j.cell.2005.02.008. PMID 15734686. S2CID 206559875.
- ↑ Mohandas E, Rajmohan V, Raghunath B (January 2009). "Neurobiology of Alzheimer's disease". Indian Journal of Psychiatry. 51 (1): 55–61. doi:10.4103/0019-5545.44908. PMC 2738403. PMID 19742193.
- ↑ Morgen K, Frölich L (April 2015). "The metabolism hypothesis of Alzheimer's disease: from the concept of central insulin resistance and associated consequences to insulin therapy". Journal of Neural Transmission. 122 (4): 499–504. doi:10.1007/s00702-015-1377-5. PMID 25673434. S2CID 21338545.
- ↑ de la Torre JC, Mussivand T (June 1993). "Can disturbed brain microcirculation cause Alzheimer's disease?". Neurological Research. 15 (3): 146–53. doi:10.1080/01616412.1993.11740127. PMID 8103579.
- ↑ Agostinho P, Cunha RA, Oliveira C (1 August 2010). "Neuroinflammation, oxidative stress and the pathogenesis of Alzheimer's disease". Current Pharmaceutical Design. 16 (25): 2766–78. doi:10.2174/138161210793176572. PMID 20698820.
- ↑ Makin S (July 2018). "The amyloid hypothesis on trial". Nature. 559 (7715): S4–S7. Bibcode:2018Natur.559S...4M. doi:10.1038/d41586-018-05719-4. PMID 30046080. S2CID 51719878.
- ↑ Davies P, Maloney AJ (December 1976). "Selective loss of central cholinergic neurons in Alzheimer's disease". Lancet. 2 (8000): 1403. doi:10.1016/S0140-6736(76)91936-X. PMID 63862. S2CID 43250282.
- 1 2 3 Mudher A, Lovestone S (January 2002). "Alzheimer's disease-do tauists and baptists finally shake hands?". Trends in Neurosciences. 25 (1): 22–6. doi:10.1016/S0166-2236(00)02031-2. PMID 11801334. S2CID 37380445.
- ↑ Schmitz C, Rutten BP, Pielen A, Schäfer S, Wirths O, Tremp G, et al. (April 2004). "Hippocampal neuron loss exceeds amyloid plaque load in a transgenic mouse model of Alzheimer's disease". The American Journal of Pathology. 164 (4): 1495–502. doi:10.1016/S0002-9440(10)63235-X. PMC 1615337. PMID 15039236.
- ↑ Gray EG, Paula-Barbosa M, Roher A (1987). "Alzheimer's disease: paired helical filaments and cytomembranes". Neuropathology and Applied Neurobiology. 13 (2): 91–110. doi:10.1111/j.1365-2990.1987.tb00174.x. PMID 3614544. S2CID 41437632.
- ↑ Williams DR (October 2006). "Tauopathies: classification and clinical update on neurodegenerative diseases associated with microtubule-associated protein tau". Internal Medicine Journal. 36 (10): 652–60. doi:10.1111/j.1445-5994.2006.01153.x. PMID 16958643. S2CID 19357113.
- ↑ Nistor M, Don M, Parekh M, Sarsoza F, Goodus M, Lopez GE, et al. (October 2007). "Alpha- and beta-secretase activity as a function of age and beta-amyloid in Down syndrome and normal brain". Neurobiology of Aging. 28 (10): 1493–506. doi:10.1016/j.neurobiolaging.2006.06.023. PMC 3375834. PMID 16904243.
- ↑ Lott IT, Head E (March 2005). "Alzheimer disease and Down syndrome: factors in pathogenesis". Neurobiology of Aging. 26 (3): 383–9. doi:10.1016/j.neurobiolaging.2004.08.005. PMID 15639317. S2CID 27716613.
- ↑ Yankner BA, Duffy LK, Kirschner DA (October 1990). "Neurotrophic and neurotoxic effects of amyloid beta protein: reversal by tachykinin neuropeptides". Science. 250 (4978): 279–82. Bibcode:1990Sci...250..279Y. doi:10.1126/science.2218531. PMID 2218531.
- ↑ Iijima K, Liu HP, Chiang AS, Hearn SA, Konsolaki M, Zhong Y (April 2004). "Dissecting the pathological effects of human Abeta40 and Abeta42 in Drosophila: a potential model for Alzheimer's disease". Proceedings of the National Academy of Sciences of the United States of America. 101 (17): 6623–8. Bibcode:2004PNAS..101.6623I. doi:10.1073/pnas.0400895101. PMC 404095. PMID 15069204.
- ↑ Gregory GC, Halliday GM (2005). "What is the dominant Abeta species in human brain tissue? A review". Neurotoxicity Research. 7 (1–2): 29–41. doi:10.1007/BF03033774. PMID 15639796. S2CID 40228398.
- ↑ Blanchard BJ, Hiniker AE, Lu CC, Margolin Y, Yu AS, Ingram VM (June 2000). "Elimination of Amyloid beta Neurotoxicity". Journal of Alzheimer's Disease. 2 (2): 137–149. doi:10.3233/JAD-2000-2214. PMID 12214104.
- ↑ Abramov AY, Canevari L, Duchen MR (December 2004). "Calcium signals induced by amyloid beta peptide and their consequences in neurons and astrocytes in culture". Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 1742 (1–3): 81–7. doi:10.1016/j.bbamcr.2004.09.006. PMID 15590058.
- ↑ Arispe N, Rojas E, Pollard HB (January 1993). "Alzheimer disease amyloid beta protein forms calcium channels in bilayer membranes: blockade by tromethamine and aluminum". Proceedings of the National Academy of Sciences of the United States of America. 90 (2): 567–71. Bibcode:1993PNAS...90..567A. doi:10.1073/pnas.90.2.567. PMC 45704. PMID 8380642.
- ↑ Barghorn S, Nimmrich V, Striebinger A, Krantz C, Keller P, Janson B, et al. (November 2005). "Globular amyloid beta-peptide oligomer - a homogenous and stable neuropathological protein in Alzheimer's disease". Journal of Neurochemistry. 95 (3): 834–47. doi:10.1111/j.1471-4159.2005.03407.x. PMID 16135089.
- ↑ Kokubo H, Kayed R, Glabe CG, Yamaguchi H (January 2005). "Soluble Abeta oligomers ultrastructurally localize to cell processes and might be related to synaptic dysfunction in Alzheimer's disease brain". Brain Research. 1031 (2): 222–8. doi:10.1016/j.brainres.2004.10.041. PMID 15649447. S2CID 54353507.
- ↑ Flagmeier P, De S, Wirthensohn DC, Lee SF, Vincke C, Muyldermans S, et al. (June 2017). "Ultrasensitive Measurement of Ca2+ Influx into Lipid Vesicles Induced by Protein Aggregates". Angewandte Chemie. 56 (27): 7750–7754. doi:10.1002/anie.201700966. PMC 5615231. PMID 28474754.
- ↑ Blanchard BJ, Chen A, Rozeboom LM, Stafford KA, Weigele P, Ingram VM (October 2004). "Efficient reversal of Alzheimer's disease fibril formation and elimination of neurotoxicity by a small molecule". Proceedings of the National Academy of Sciences of the United States of America. 101 (40): 14326–32. Bibcode:2004PNAS..10114326B. doi:10.1073/pnas.0405941101. PMC 521943. PMID 15388848.
- ↑ Porat Y, Abramowitz A, Gazit E (January 2006). "Inhibition of amyloid fibril formation by polyphenols: structural similarity and aromatic interactions as a common inhibition mechanism". Chemical Biology & Drug Design. 67 (1): 27–37. doi:10.1111/j.1747-0285.2005.00318.x. PMID 16492146.
- ↑ Kanapathipillai M, Lentzen G, Sierks M, Park CB (August 2005). "Ectoine and hydroxyectoine inhibit aggregation and neurotoxicity of Alzheimer's beta-amyloid". FEBS Letters. 579 (21): 4775–80. doi:10.1016/j.febslet.2005.07.057. PMID 16098972.
- 1 2 Himeno E, Ohyagi Y, Ma L, Nakamura N, Miyoshi K, Sakae N, et al. (February 2011). "Apomorphine treatment in Alzheimer mice promoting amyloid-β degradation". Annals of Neurology. 69 (2): 248–56. doi:10.1002/ana.22319. PMID 21387370. S2CID 242138.
- ↑ Lashuel HA, Hartley DM, Balakhaneh D, Aggarwal A, Teichberg S, Callaway DJ (November 2002). "New class of inhibitors of amyloid-beta fibril formation. Implications for the mechanism of pathogenesis in Alzheimer's disease". The Journal of Biological Chemistry. 277 (45): 42881–90. doi:10.1074/jbc.M206593200. PMID 12167652.
- ↑ Lee KH, Shin BH, Shin KJ, Kim DJ, Yu J (March 2005). "A hybrid molecule that prohibits amyloid fibrils and alleviates neuronal toxicity induced by beta-amyloid (1-42)". Biochemical and Biophysical Research Communications. 328 (4): 816–23. doi:10.1016/j.bbrc.2005.01.030. PMID 15707952.
- ↑ Espeseth AS, Xu M, Huang Q, Coburn CA, Jones KL, Ferrer M, et al. (May 2005). "Compounds that bind APP and inhibit Abeta processing in vitro suggest a novel approach to Alzheimer disease therapeutics". The Journal of Biological Chemistry. 280 (18): 17792–7. doi:10.1074/jbc.M414331200. PMID 15737955.
- ↑ Moreno H, Yu E, Pigino G, Hernandez AI, Kim N, Moreira JE, et al. (April 2009). "Synaptic transmission block by presynaptic injection of oligomeric amyloid beta". Proceedings of the National Academy of Sciences of the United States of America. 106 (14): 5901–6. Bibcode:2009PNAS..106.5901M. doi:10.1073/pnas.0900944106. PMC 2659170. PMID 19304802.
- ↑ Pigino G, Morfini G, Atagi Y, Deshpande A, Yu C, Jungbauer L, et al. (April 2009). "Disruption of fast axonal transport is a pathogenic mechanism for intraneuronal amyloid beta". Proceedings of the National Academy of Sciences of the United States of America. 106 (14): 5907–12. Bibcode:2009PNAS..106.5907P. doi:10.1073/pnas.0901229106. PMC 2667037. PMID 19321417.
- ↑ Kinney JW, Bemiller SM, Murtishaw AS, Leisgang AM, Salazar AM, Lamb BT (January 2018). "Inflammation as a central mechanism in Alzheimer's disease". Alzheimer's & Dementia. 4 (1): 575–590. doi:10.1016/j.trci.2018.06.014. PMC 6214864. PMID 30406177.
- ↑ Griffin WS, Sheng JG, Roberts GW, Mrak RE (March 1995). "Interleukin-1 expression in different plaque types in Alzheimer's disease: significance in plaque evolution". Journal of Neuropathology and Experimental Neurology. 54 (2): 276–81. doi:10.1097/00005072-199503000-00014. PMID 7876895. S2CID 33277264.
- ↑ Griffin WS, Stanley LC, Ling C, White L, MacLeod V, Perrot LJ, et al. (October 1989). "Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease". Proceedings of the National Academy of Sciences of the United States of America. 86 (19): 7611–5. Bibcode:1989PNAS...86.7611G. doi:10.1073/pnas.86.19.7611. PMC 298116. PMID 2529544.
- ↑ Gomez-Nicola D, Boche D (December 2015). "Post-mortem analysis of neuroinflammatory changes in human Alzheimer's disease". Alzheimer's Research & Therapy. 7 (1): 42. doi:10.1186/s13195-015-0126-1. PMC 4405851. PMID 25904988.
- ↑ Knezevic D, Mizrahi R (January 2018). "Molecular imaging of neuroinflammation in Alzheimer's disease and mild cognitive impairment". Progress in Neuro-Psychopharmacology & Biological Psychiatry. 80 (Pt B): 123–131. doi:10.1016/j.pnpbp.2017.05.007. PMID 28533150. S2CID 31181575.
- ↑ McGeer PL, McGeer EG (October 2013). "The amyloid cascade-inflammatory hypothesis of Alzheimer disease: implications for therapy". Acta Neuropathologica. 126 (4): 479–97. doi:10.1007/s00401-013-1177-7. PMID 24052108. S2CID 32212325.
- ↑ Brothers HM, Gosztyla ML, Robinson SR (2018-04-25). "The Physiological Roles of Amyloid-β Peptide Hint at New Ways to Treat Alzheimer's Disease". Frontiers in Aging Neuroscience. 10: 118. doi:10.3389/fnagi.2018.00118. PMC 5996906. PMID 29922148.
- ↑ Kreisl WC (July 2017). "Discerning the relationship between microglial activation and Alzheimer's disease". Brain. 140 (7): 1825–1828. doi:10.1093/brain/awx151. PMID 29177498.
- ↑ Kinney JW, Bemiller SM, Murtishaw AS, Leisgang AM, Salazar AM, Lamb BT (January 2018). "Inflammation as a central mechanism in Alzheimer's disease". Alzheimer's & Dementia. 4 (1): 575–590. doi:10.1016/j.trci.2018.06.014. PMC 6214864. PMID 30406177.
- ↑ Agostinho P, Cunha RA, Oliveira C (2010-08-01). "Neuroinflammation, oxidative stress and the pathogenesis of Alzheimer's disease". Current Pharmaceutical Design. 16 (25): 2766–78. doi:10.2174/138161210793176572. PMID 20698820.
- ↑ Wang WY, Tan MS, Yu JT, Tan L (June 2015). "Role of pro-inflammatory cytokines released from microglia in Alzheimer's disease". Annals of Translational Medicine. 3 (10): 136. doi:10.3978/j.issn.2305-5839.2015.03.49. PMC 4486922. PMID 26207229.
- ↑ Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, et al. (2000). "Inflammation and Alzheimer's disease". Neurobiology of Aging. 21 (3): 383–421. doi:10.1016/s0197-4580(00)00124-x. PMC 3887148. PMID 10858586.
- ↑ Tuppo EE, Arias HR (February 2005). "The role of inflammation in Alzheimer's disease". The International Journal of Biochemistry & Cell Biology. 37 (2): 289–305. doi:10.1016/j.biocel.2004.07.009. PMID 15474976.
- ↑ Meraz-Ríos MA, Toral-Rios D, Franco-Bocanegra D, Villeda-Hernández J, Campos-Peña V (2013). "Inflammatory process in Alzheimer's Disease". Frontiers in Integrative Neuroscience. 7: 59. doi:10.3389/fnint.2013.00059. PMC 3741576. PMID 23964211.
- ↑ Edlund C, Söderberg M, Kristensson K (July 1994). "Isoprenoids in aging and neurodegeneration". Neurochemistry International. 25 (1): 35–8. doi:10.1016/0197-0186(94)90050-7. PMID 7950967. S2CID 34009482.
- ↑ Edlund C, Söderberg M, Kristensson K (July 1994). "Isoprenoids in aging and neurodegeneration". Neurochemistry International. 25 (1): 35–8. doi:10.1016/0197-0186(94)90050-7. PMID 7950967. S2CID 34009482.
- ↑ Sviderskii VL, Khovanskikh AE, Rozengart EV, Moralev SN, Yagodina OV, Gorelkin VS, et al. (2006). "A comparative study of the effect of the polyprenol preparation ropren from coniferous plants on the key enzymes of the cholinergic and monoaminergic types of nervous transmission". Doklady. Biochemistry and Biophysics (in русский). 408: 148–51. doi:10.1134/S1607672906030112. PMID 16913416. S2CID 12431221.
- Lay summary in: "Biotechnology News".
{{cite journal}}: Cite journal requires|journal=(help)
- Lay summary in: "Biotechnology News".
- ↑ Fedotova J, Soultanov V, Nikitina T, Roschin V, Ordayn N (March 2012). "Ropren(®) is a polyprenol preparation from coniferous plants that ameliorates cognitive deficiency in a rat model of beta-amyloid peptide-(25-35)-induced amnesia". Phytomedicine. 19 (5): 451–6. doi:10.1016/j.phymed.2011.09.073. PMID 22305275.
- ↑ Cunnane S, Nugent S, Roy M, Courchesne-Loyer A, Croteau E, Tremblay S, et al. (January 2011). "Brain fuel metabolism, aging, and Alzheimer's disease". Nutrition. 27 (1): 3–20. doi:10.1016/j.nut.2010.07.021. PMC 3478067. PMID 21035308.
- 1 2 3 Costantini LC, Barr LJ, Vogel JL, Henderson ST (December 2008). "Hypometabolism as a therapeutic target in Alzheimer's disease". BMC Neuroscience. 9 Suppl 2 (Suppl 2): S16. doi:10.1186/1471-2202-9-s2-s16. PMC 2604900. PMID 19090989.
- ↑ Hoyer S (June 1992). "Oxidative energy metabolism in Alzheimer brain. Studies in early-onset and late-onset cases". Molecular and Chemical Neuropathology. 16 (3): 207–24. doi:10.1007/bf03159971. PMID 1418218.
- ↑ Small GW, Ercoli LM, Silverman DH, Huang SC, Komo S, Bookheimer SY, et al. (May 2000). "Cerebral metabolic and cognitive decline in persons at genetic risk for Alzheimer's disease". Proceedings of the National Academy of Sciences of the United States of America. 97 (11): 6037–42. Bibcode:2000PNAS...97.6037S. doi:10.1073/pnas.090106797. PMC 18554. PMID 10811879.
- 1 2 de Leon MJ, Ferris SH, George AE, Christman DR, Fowler JS, Gentes C, et al. (1983). "Positron emission tomographic studies of aging and Alzheimer disease". AJNR. American Journal of Neuroradiology. 4 (3): 568–71. PMC 8334899. PMID 6410799.
- ↑ Meier-Ruge W, Bertoni-Freddari C, Iwangoff P (1994). "Changes in brain glucose metabolism as a key to the pathogenesis of Alzheimer's disease". Gerontology. 40 (5): 246–52. doi:10.1159/000213592. PMID 7959080.
- 1 2 Reiman EM, Chen K, Alexander GE, Caselli RJ, Bandy D, Osborne D, et al. (January 2004). "Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer's dementia". Proceedings of the National Academy of Sciences of the United States of America. 101 (1): 284–9. Bibcode:2003PNAS..101..284R. doi:10.1073/pnas.2635903100. PMC 314177. PMID 14688411.
- ↑ Corder EH, Jelic V, Basun H, Lannfelt L, Valind S, Winblad B, Nordberg A (March 1997). "No difference in cerebral glucose metabolism in patients with Alzheimer disease and differing apolipoprotein E genotypes". Archives of Neurology. 54 (3): 273–7. doi:10.1001/archneur.1997.00550150035013. PMID 9074396.
- ↑ "Diminished cerebral glucose metabolism: A key pathology in Alzheimer's disease" (PDF). Archived from the original (PDF) on 15 October 2013. Retrieved 9 October 2013.
- 1 2 3 4 De Felice FG (February 2013). "Alzheimer's disease and insulin resistance: translating basic science into clinical applications". The Journal of Clinical Investigation. 123 (2): 531–9. doi:10.1172/JCI64595. PMC 3561831. PMID 23485579.
- ↑ De Felice FG, Vieira MN, Bomfim TR, Decker H, Velasco PT, Lambert MP, et al. (February 2009). "Protection of synapses against Alzheimer's-linked toxins: insulin signaling prevents the pathogenic binding of Abeta oligomers". Proceedings of the National Academy of Sciences of the United States of America. 106 (6): 1971–6. Bibcode:2009PNAS..106.1971D. doi:10.1073/pnas.0809158106. PMC 2634809. PMID 19188609.
- ↑ Herzer S, Meldner S, Rehder K, Gröne HJ, Nordström V (September 2016). "Lipid microdomain modification sustains neuronal viability in models of Alzheimer's disease". Acta Neuropathologica Communications. 4 (1): 103. doi:10.1186/s40478-016-0354-z. PMC 5027102. PMID 27639375.
- ↑ Wan Q, Xiong ZG, Man HY, Ackerley CA, Braunton J, Lu WY, et al. (August 1997). "Recruitment of functional GABA(A) receptors to postsynaptic domains by insulin". Nature. 388 (6643): 686–90. Bibcode:1997Natur.388..686W. doi:10.1038/41792. PMID 9262404. S2CID 4383461.
- ↑ Saraiva LM, Seixas da Silva GS, Galina A, da-Silva WS, Klein WL, Ferreira ST, De Felice FG (December 2010). "Amyloid-β triggers the release of neuronal hexokinase 1 from mitochondria". PLOS ONE. 5 (12): e15230. Bibcode:2010PLoSO...515230S. doi:10.1371/journal.pone.0015230. PMC 3002973. PMID 21179577.
- ↑ Craft S (June 2012). "Alzheimer disease: Insulin resistance and AD--extending the translational path". Nature Reviews. Neurology. 8 (7): 360–2. doi:10.1038/nrneurol.2012.112. PMID 22710630. S2CID 31213610. Archived from the original on 2022-01-11. Retrieved 2021-11-23.
- ↑ de la Monte SM (January 2012). "Brain insulin resistance and deficiency as therapeutic targets in Alzheimer's disease". Current Alzheimer Research. 9 (1): 35–66. doi:10.2174/156720512799015037. PMC 3349985. PMID 22329651.
- ↑ Liu Z, Zhou T, Ziegler AC, Dimitrion P, Zuo L (2017). "Oxidative Stress in Neurodegenerative Diseases: From Molecular Mechanisms to Clinical Applications". Oxidative Medicine and Cellular Longevity. 2017: 2525967. doi:10.1155/2017/2525967. PMC 5529664. PMID 28785371.
- 1 2 Bonda DJ, Wang X, Lee HG, Smith MA, Perry G, Zhu X (April 2014). "Neuronal failure in Alzheimer's disease: a view through the oxidative stress looking-glass". Neuroscience Bulletin. 30 (2): 243–52. doi:10.1007/s12264-013-1424-x. PMC 4097013. PMID 24733654.
- ↑ Wang J, Xiong S, Xie C, Markesbery WR, Lovell MA (May 2005). "Increased oxidative damage in nuclear and mitochondrial DNA in Alzheimer's disease". Journal of Neurochemistry. 93 (4): 953–62. doi:10.1111/j.1471-4159.2005.03053.x. PMID 15857398.
- ↑ Wang J, Markesbery WR, Lovell MA (February 2006). "Increased oxidative damage in nuclear and mitochondrial DNA in mild cognitive impairment". Journal of Neurochemistry. 96 (3): 825–32. doi:10.1111/j.1471-4159.2005.03615.x. PMID 16405502.
- ↑ Soheili-Nezhad S (2017). "Alzheimer's disease: the large gene instability hypothesis". bioRxiv. doi:10.1101/189712.
- ↑ Smith DI, Zhu Y, McAvoy S, Kuhn R (January 2006). "Common fragile sites, extremely large genes, neural development and cancer". Cancer Letters. 232 (1): 48–57. doi:10.1016/j.canlet.2005.06.049. PMID 16221525.
- ↑ Vilenchik MM, Knudson AG (October 2003). "Endogenous DNA double-strand breaks: production, fidelity of repair, and induction of cancer". Proceedings of the National Academy of Sciences of the United States of America. 100 (22): 12871–6. Bibcode:2003PNAS..10012871V. doi:10.1073/pnas.2135498100. PMC 240711. PMID 14566050.
- ↑ Madabhushi R, Gao F, Pfenning AR, Pan L, Yamakawa S, Seo J, et al. (June 2015). "Activity-Induced DNA Breaks Govern the Expression of Neuronal Early-Response Genes". Cell. 161 (7): 1592–605. doi:10.1016/j.cell.2015.05.032. PMC 4886855. PMID 26052046.
- 1 2 Thadathil N, Delotterie DF, Xiao J, Hori R, McDonald MP, Khan MM (January 2021). "DNA Double-Strand Break Accumulation in Alzheimer's Disease: Evidence from Experimental Models and Postmortem Human Brains". Molecular Neurobiology. 58 (1): 118–131. doi:10.1007/s12035-020-02109-8. PMID 32895786. S2CID 221541995.
- ↑ Shanbhag NM, Evans MD, Mao W, Nana AL, Seeley WW, Adame A, et al. (May 2019). "Early neuronal accumulation of DNA double strand breaks in Alzheimer's disease". Acta Neuropathologica Communications. 7 (1): 77. doi:10.1186/s40478-019-0723-5. PMC 6524256. PMID 31101070.
- ↑ Shackelford DA (April 2006). "DNA end joining activity is reduced in Alzheimer's disease". Neurobiology of Aging. 27 (4): 596–605. doi:10.1016/j.neurobiolaging.2005.03.009. PMID 15908050. S2CID 7327609.