Crouzonodermoskeletal syndrome
| Crouzonodermoskeletal syndrome | |
|---|---|
| Other names: Crouzon syndrome with acanthosis nigricans | |
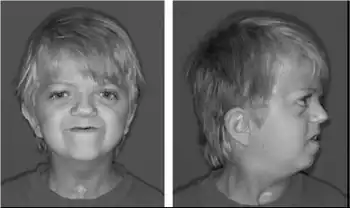 | |
| Craniofacial and skin findings in Crouzonodermoskeletal syndrome-midfacial hypoplasia , periorbital and perioral acanthosis nigricans. This individual was found to have the FGFR3 (A319E) mutation | |
| Specialty | Medical genetics |
Crouzonodermoskeletal syndrome is a disorder characterized by the premature joining of certain bones of the skull (craniosynostosis) during development and a skin condition called acanthosis nigricans.[1]
Signs and symptoms
Some of the signs and symptoms of Crouzonodermoskeletal syndrome are similar to those seen with Crouzon syndrome. They include prematurely fused skull bones, which affect the shape of the head and face; wide-set, bulging eyes due to shallow eye sockets; eyes that do not point in the same direction (strabismus); a small, beaked nose; and an underdeveloped upper jaw. People with these conditions are generally of normal intelligence.[2]
Genetics
Mutations in the FGFR3 gene cause Crouzonodermoskeletal syndrome. The protein made by the FGFR3 gene is a receptor that plays a role in the development and maintenance of bone and brain tissue. Researchers do not know how a mutation in FGFR3 leads to the characteristic features of this disorder, but changes in the receptor appear to disrupt the normal development of bones in the skull and affect skin pigmentation.[3]
This condition is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.
In some cases, an affected person inherits the mutation from one affected parent. Other cases may result from new mutations in the gene. These cases occur in people with no history of the disorder in their family.
Diagnosis
Differential diagnosis
Several features distinguish Crouzonodermoskeletal syndrome from Crouzon syndrome. People with Crouzonodermoskeletal syndrome have acanthosis nigricans, a skin condition characterized by thick, dark, velvety skin in body folds and creases, including the neck and underarms. In addition, subtle changes may be seen in the bones of the spine (vertebrae). Noncancerous growths called cementomas may develop in the jaw during young adulthood.Crouzonodermoskeletal syndrome is rare; the condition is seen in about 1 per million people.
Treatment
In terms of management, affected individuals will benefit from a multidisciplinary medical team, which would include eventually some degree of surgery[4]
References
- ↑ Herman, TE; Sargar, K; Siegel, MJ (Feb 2014). "Crouzono-dermo-skeletal syndrome, Crouzon syndrome with acanthosis nigricans syndrome". Journal of Perinatology. 34 (2): 164–5. doi:10.1038/jp.2013.139. PMID 24476664. S2CID 38093032.
- ↑ Schweitzer DN, Graham JM Jr, Lachman RS, Jabs EW, Okajima K, Przylepa KA, Shanske A, Chen K, Neidich JA, Wilcox WR (2001). "Subtle radiographic findings of achondroplasia in patients with Crouzon syndrome with acanthosis nigricans due to an Ala391Glu substitution in FGFR3". Am J Med Genet. 98 (1): 75–91. doi:10.1002/1096-8628(20010101)98:1<75::AID-AJMG1010>3.0.CO;2-6. PMID 11426459.
- ↑ Vajo, Zoltan, Francomano CA, Wilkin DJ (2000). "The molecular and genetic basis of fibroblast growth factor receptor 3 disorders: the achondroplasia family of skeletal dysplasias, Muenke craniosynostosis, and Crouzon syndrome with acanthosis nigricans". Endocr Rev. 21 (1): 23–39. doi:10.1210/edrv.21.1.0387. PMID 10696568. Archived from the original on 2020-12-05. Retrieved 2022-06-13.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - ↑ Wenger, Tara; Miller, Danny; Evans, Kelly (1993). "FGFR Craniosynostosis Syndromes Overview". GeneReviews®. University of Washington, Seattle. Archived from the original on 23 January 2021. Retrieved 25 June 2022.
- Cohen MM Jr (1999). "Let's call it "Crouzonodermoskeletal syndrome" so we won't be prisoners of our own conventional terminology". Am J Med Genet. 84 (1): 74. doi:10.1002/(SICI)1096-8628(19990507)84:1<74::AID-AJMG14>3.0.CO;2-R. PMID 10213050.
External links
| Classification | |
|---|---|
| External resources |
|