Dermatofibrosarcoma protuberans, fibrosarcomatous
| Dermatofibrosarcoma protuberans, fibrosarcomatous | |
|---|---|
| Other names: Fibrosarcomatous dermatofibrosarcoma protuberans | |
| Specialty | Dermatology, Oncology, Pathology, Surgical oncology |
| Symptoms | Painless mass in the dermis |
| Complications | Multiple recurrences and metastases |
| Usual onset | Adults 30-59 years/old |
| Causes | Formation of a COL1A1-PDGFB fusion gene |
| Treatment | Surgical excision, radiotherapy, protein kinase inhibitors |
| Prognosis | Guarded |
| Frequency | Rare |
| Deaths | Uncommon |
Dermatofibrosarcoma protuberans, fibrosarcomatous (DFSP-FS), also termed fibrosarcomatous dermatofibrosarcoma protuberans, is a rare type of tumor located in the dermis (i.e. layer of the skin below the epidermis).[1] DFSP-FS tumors have been viewed as: 1) a more aggressive form of the dermatofibrosarcoma protuberans (DFSP) tumors because they have areas that resemble and tend to behave like malignant fibrosarcomas[2] or 2) as a distinctly different tumor than DFSP.[3] DFSP-FS tumors are related to DFSP.[4] For example, surgically removed DFSP tumors often recur with newly developed fibrobosarcoma-like areas.[5] Nonetheless, the World Health Organization (WHO), 2020, classified DFSP and DFSP-FS as different tumors with DFSP being in the category of benign and DFSP-FS in the category of rarely metastasizing fibroblastic and myofibroblastic tumors.[6] This article follows the WHO classification: the 5-15% of DFSP tumors that have any areas of fibrosarcomatous microscopic histopathology[3] are here considered DFSP-FS rather than DFSP tumors.
DFSP tumors typically consist of bland-appearing, slowly proliferating, spindle-shaped cells arranged in a monotonous cartwheel or whorled pattern. DFSP-FS tumors consist of less bland-appearing spindle-shaped cells that are arranged in fascicular (i.e. bundled, smooth muscle-like) or herringbone-like patterns, have large, vesicular, misshaped nuclei, and are rapidly proliferating; these DFSP-FS areas are typically but not always admixed with DFSP areas.[7] Various studies find that DFSP-FS tumors have higher rates of recurrence after surgical removal than DFSP tumors and may metastasize (i.e. spread to distant tissues).[2][8][9][10]
The tumor cells in DFSP and DFSP-FS harbor one or more fusion gene mutations, i.e. mutations that merge two previously independent genes. The COL1A1-PDGFB fusion gene is the most common fusion gene found in both tumor types.[11][12] However, DFSP-FS tumor cells have higher copy numbers of the COL1A1-PDGFB fusion gene than do DFSP tumors.[7][12]
Localized DFSP-FS tumors are typically treated by wide surgical excision in order to reduce the high recurrence rates developing when these tumors' cells are not completely removed. Adjuvant therapy (i.e. therapy given in addition to the primary or initial therapy in order to maximize its effectiveness) consisting of radiation therapy and/or drugs (i.e. protein kinase inhibitors that block the effects of the COL1A1-PDGFB fusion gene) may be added to the treatment regimen in cases where a tumor cannot be fully removed and in virtually all cases where the tumor has metastasized.[8]
Signs and symptoms
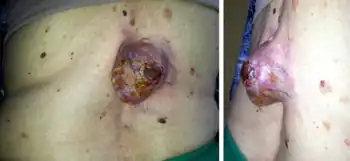
DFSP-FS tumors are diagnosed primarily in adults aged 30–59 years old;[13] DFSP-SF is rare in children[14] with only 12 cases been reported in individuals 8–20 years old as of 2021.[15] The initial lesions in DFSP and DFSP-FS typically begin as small, single, painless, violet or pink cutaneous nodules or plaques (i.e. elevated areas of skin 2 cm. or more in diameter) located in the trunk, abdomen, limbs, head, or neck.[16] They may be stable or grow slowly for years[13] but most cases, ~81% in one study,[9] then begin to grow faster and may reach large sizes (e.g. 40 cm.).[2] In general, there are no differences between DFSP-FS and DFSP in the lesion size or inflicted individuals' sex at presentation.[9]
DFSP-FS tumors are often diagnosed in tumors that have recurred at the sites where DFSP tumors were surgically removed one or more times. In general, they are more rapidly growing and invasive (e.g. rare DFSP-FS dermal tumors in the chest or abdominal skin have respectively invaded the thoracic[17] or abdominal cavities[18]) than DFSP tumors. About 10[13] to 15%[1] of individuals initially or subsequently present with metastases,[2] most commonly in the lung, less commonly in the regional lymph nodes draining the site of the primary dermal tumor,[13] or, rarely, in the liver, kidney, soft tissue sites outside of the primary dermal tumor's areas,[3] mediastinum, or brain.[19]
Pathology
The microscopic histopathologic findings in hematoxylin and eosin-stained DFSP tumor tissues typically show bland, uniform, spindle-shaped tumor cells in the dermis (and often the adjacent subcutaneous fat tissues); these cells are arranged in characteristic cartwheel or whorled patterns.[20] The tumors cells express CD34 protein in 92% to 100% of cases,[13] nestin protein in 95% of cases,[13] and S100 protein and melanocyte protein PMEL protein in some cases.[8]
DFSP-FS tumors have a DFSP-FS histopathology in 5%[16] to 100%[7] of their total tissue areas; the DFSP-FS areas consist of densely packed spindle-shaped cells arranged in smooth muscle-like bundled[8] or herringbone[13] patterns. The spindle-shaped cells have abnormally large nuclear vesicles, large, misshaped nuclei, and rapid proliferation rates.[1] In almost all cases, however, DFSP-FS have at least some DFSP areas that abruptly or gradually transition into DFSP-FS areas or vice versa.[16] DFSP-FS tumors tend to be more deeply invasive than DFSP tumors.[8][17][18] In 45% of cases, CD34 protein is absent or greatly reduced in DFSP-FS tumor cells compared to DFSP tumor cells.[8] Like DFSP tumor cells, DFSP-FS tumor cells commonly express nestin protein.[13]
Chromosome and gene abnormalities
DFSP and DFSP-FS tumor cells contain at least one COL1A1-PDGFB fusion gene. A fusion gene is a hybrid gene formed by an abnormal merger between two previously independent genes. The COL1A1 gene, which directs production of collagen, type I, alpha 1 protein, is normally located in band 21.33 on the long (or "q") arm of chromosome 17.[21] The PDGFB gene, which directs production of platelet-derived growth factor subunit B (PDGFβ) protein, is normally located in band 13.1 on the q arm of chromosome 22. The COL1A1 and PDGFB genes in DFSP-FS and DFSP are most commonly merged as result of the formation of a small supernumerary ring chromosome, i.e. an extra ring-shaped chromosome that in this case juxtapositions the COL1A1 and PDGFB genes. Less commonly, this fusion gene forms as a result of a translocation which merges the two genes sites onto either chromosome 17 or 22. In both casse, the COL1A1-PDGFB fusion genes direct the overproduction of fully active PDGFβ proteins.[1] The overproduced PDGFβ proteins bind to platelet-derived growth factor receptors to promote these receptors' intrinsic tyrosine kinase activity and thereby to overstimulate mitogen-activated protein kinases, PI3K/AKT/mTOR, and other cell signaling pathways which promote the growth, proliferation, and prolonged survival of their parent cells. These events are considered to underlie the development and/or progression of DFSP and DFSP-FS tumors.[8][22] While the cells of both tumor types tend to have multiple copies of the COL1A1-PDGFB fusion gene, DFSP-FS tumor cells tend to have more copies of them than DFSP tumor cells. This difference may contribute to the more aggressive behavior of DFSP-FS.[1]
While the COL1A1-PDGFB fusion gene is present in >90% of DFSP-FS and DFSP cases, recent studies have also found the COL6A3-PDGFD and EMILIN2-PDGFD (EMILIN2 is the gene for elastin microfibril interfacer 2 protein[23]) fusions genes in <2% of DFSP-FS or DFSP cases, the TNC-PDGFD fusion gene in one case of DFSP-FS,[11] the COL1A2-PDGFB fusion gene in one case of DFSP,[24] and the CSPG2-PTK2B fusion gene in one case of DFSP-FS.[8] Further studies are needed to determine the latter five fusion genes' prevalence in, and contribution to the development and/or progression of, DFSP and DFSP-FS.
Diagnosis
The diagnosis of DFSP-FS depends on finding a dermal tumor with characteristic areas consisting of spindle-shaped cells with atypical, vesicular nuclei aligned in smooth muscle-like bundled[8] or herringbone patterns.[13] The spindle-shaped cells are often rapidly growing as indicated by their relatively high mitotic index.[1] These sites form from 5%[16] to 100%[7] of the tumor with any remaining sites consisting of DFSP lesions (i.e. bland, uniform, relatively slowly proliferating spindle-shaped tumor cells arranged in a characteristic cartwheel or whorled pattern).[20] The diagnosis of DFSP-FS is strongly supported by finding that the tumors' cells express the COL1A1-PDGFB fusion gene; a finding that these tumor cells do not express or only weakly express CD34 protein strongly indicates that it is a DFSP-FS rather than a DFSP tumor.[8]
Individuals with DFSP-FS should have radiographic,[20] CT scan, and/or magnetic resonance studies of the chest[8] as well as ultrasound and/or CT scan studies of the abdomen and the lymph nodes which drain the tumor sites in order to determine if metastatic disease is present.[20]
Treatment
Localized DFSP-FS tumors are typically treated by wide local excision or Mohs microscopic surgery in order to avoid the high recurrence rates that occur when these tumors' cells are left behind.[8][13] Cases in which post-surgical analysis indicate that tumor cells have been left behind are further treated with radiotherapy directed at the surgical site.[2] Tumors located on a finger or toe, which trend to have a high and rapidly developing recurrence rate, may be treated by partial or total amputation.[16] Tumors that are only partly resectable have been treated with partial resection combined with radiotherapy, drugs that inhibit the tyrosine kinase on the platelet-derived growth factor receptors stimulated by DFSP-FS (in cases where the tumor cells express the COL1A1-PDGFB fusion gene),[8] and/or, in rare cases, chemotherapy regimens.[7][17][18] Large tumors in cosmetically sensitive locations, tumors that are completely unresectable, and tumors associated with metastatic tumors have likewise been treated with radiotherapy, tyrosine kinase inhibitors,[13] and/or chemotherapy.[17][18]
Among the tyrosine kinase inhibitors used to treat DFSP-FS, imatinib (which is approved by the FDA for treating DFSP[25]) has produced 73% partial and 90% stable disease responses. Sunitinib, which inhibits various types of tyrosine kinases, has been used as a second-line agent for the treatment of imatinib-resistant tumors: 30 patients who developed resistance to imatinib had an 80% overall rate of disease control in response to this drug. Sorafenib, another inhibitor of various tyrosine kinases, has also been suggested as a potential treatment for DFSP-FS based on a single report describing s patient who responded to sorafenib after failing to respond to imatinib.[13] Chemotherapy regimens used to treat DFSP-FS include doxorubicin plus ifosfamide together [19] or combined with an aromatase inhibitor[17] and doxorubicin and ifosfamide combined with dacarbazine.[18] These chemotherapy regimens had only short-lived, marginal, or no apparent effects in these cases.
Follow-ups in patients treated for DFSP-FS are aimed at the early detection of local recurrences and metastatic disease.[26] One study recommends that the treatment regimen should include physical examinations and imaging studies (see Diagnosis section) that are routinely conducted following initial therapy every 6 months for 5 years and yearly thereafter.[20]
Prognosis
Retrospective reviews of patients treated for DFSP-FS find average 5-year recurrence-free survival rates of 42%-52% with a 10%-15% risk of having or developing metastatic disease.[13] In an earlier (2014) retrospective review of 225 cases treated for DFSP-FS, 29.8% developed post-surgical recurrent disease (median time after surgery: 0.9-7.8 years), 14.4% had or developed metastatic disease, and 14.7% died from their disease (median time after surgery: 14.7 years).[3]
References
- 1 2 3 4 5 6 Baranov E, Hornick JL (March 2020). "Soft Tissue Special Issue: Fibroblastic and Myofibroblastic Neoplasms of the Head and Neck". Head and Neck Pathology. 14 (1): 43–58. doi:10.1007/s12105-019-01104-3. PMC 7021862. PMID 31950474.
- 1 2 3 4 5 Verma H, Sehgal K, Panchal KB, Chakraborty S, Biswas B, Mukherjee G, Midha D, Biswas G (March 2020). "Presentation and Management of Dermatofibrosarcoma Protuberans: a Single Center Protocol". Indian Journal of Surgical Oncology. 11 (1): 35–40. doi:10.1007/s13193-019-01007-3. PMC 7064730. PMID 32205967.
- 1 2 3 4 Liang CA, Jambusaria-Pahlajani A, Karia PS, Elenitsas R, Zhang PD, Schmults CD (October 2014). "A systematic review of outcome data for dermatofibrosarcoma protuberans with and without fibrosarcomatous change". Journal of the American Academy of Dermatology. 71 (4): 781–6. doi:10.1016/j.jaad.2014.03.018. PMID 24755121.
- ↑ Braswell DS, Ayoubi N, Motaparthi K, Walker A (April 2020). "Dermatofibrosarcoma protuberans with features of giant cell fibroblastoma in an adult". Journal of Cutaneous Pathology. 47 (4): 317–320. doi:10.1111/cup.13601. PMID 32163628. S2CID 212691248.
- ↑ Jha P, Moosavi C, Fanburg-Smith JC (April 2007). "Giant cell fibroblastoma: an update and addition of 86 new cases from the Armed Forces Institute of Pathology, in honor of Dr. Franz M. Enzinger". Annals of Diagnostic Pathology. 11 (2): 81–8. doi:10.1016/j.anndiagpath.2006.12.010. PMID 17349565.
- ↑ Sbaraglia M, Bellan E, Dei Tos AP (April 2021). "The 2020 WHO Classification of Soft Tissue Tumours: news and perspectives". Pathologica. 113 (2): 70–84. doi:10.32074/1591-951X-213. PMC 8167394. PMID 33179614.
- 1 2 3 4 5 Mentzel T, Beham A, Katenkamp D, Dei Tos AP, Fletcher CD (May 1998). "Fibrosarcomatous ("high-grade") dermatofibrosarcoma protuberans: clinicopathologic and immunohistochemical study of a series of 41 cases with emphasis on prognostic significance". The American Journal of Surgical Pathology. 22 (5): 576–87. doi:10.1097/00000478-199805000-00009. PMID 9591728.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 Hao X, Billings SD, Wu F, Stultz TW, Procop GW, Mirkin G, Vidimos AT (June 2020). "Dermatofibrosarcoma Protuberans: Update on the Diagnosis and Treatment". Journal of Clinical Medicine. 9 (6): 1752. doi:10.3390/jcm9061752. PMC 7355835. PMID 32516921.
- 1 2 3 Li Y, Liang J, Xu X, Jiang X, Wang C, Chen S, Xiang B, Ji Y (January 2021). "Clinicopathological features of fibrosarcomatous dermatofibrosarcoma protuberans and the construction of a back-propagation neural network recognition model". Orphanet Journal of Rare Diseases. 16 (1): 48. doi:10.1186/s13023-021-01698-4. PMC 7836157. PMID 33499900.
- ↑ Qu Q, Xuan W, Fan GH (January 2015). "Roles of resolvins in the resolution of acute inflammation". Cell Biology International. 39 (1): 3–22. doi:10.1002/cbin.10345. PMID 25052386. S2CID 10160642.
- 1 2 Chen Y, Shi YZ, Feng XH, Wang XT, He XL, Zhao M (July 2021). "Novel TNC-PDGFD fusion in fibrosarcomatous dermatofibrosarcoma protuberans: a case report". Diagnostic Pathology. 16 (1): 63. doi:10.1186/s13000-021-01123-1. PMC 8276425. PMID 34256767.
- 1 2 Abbott JJ, Erickson-Johnson M, Wang X, Nascimento AG, Oliveira AM (November 2006). "Gains of COL1A1-PDGFB genomic copies occur in fibrosarcomatous transformation of dermatofibrosarcoma protuberans". Modern Pathology. 19 (11): 1512–8. doi:10.1038/modpathol.3800695. PMID 16980946. S2CID 276608.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 Allen A, Ahn C, Sangüeza OP (October 2019). "Dermatofibrosarcoma Protuberans". Dermatologic Clinics. 37 (4): 483–488. doi:10.1016/j.det.2019.05.006. PMID 31466588. S2CID 201672710.
- ↑ Terrier-Lacombe MJ, Guillou L, Maire G, Terrier P, Vince DR, de Saint Aubain Somerhausen N, Collin F, Pedeutour F, Coindre JM (January 2003). "Dermatofibrosarcoma protuberans, giant cell fibroblastoma, and hybrid lesions in children: clinicopathologic comparative analysis of 28 cases with molecular data--a study from the French Federation of Cancer Centers Sarcoma Group". The American Journal of Surgical Pathology. 27 (1): 27–39. doi:10.1097/00000478-200301000-00004. PMID 12502925. S2CID 34359313.
- ↑ Chicaud M, Frassati-Biaggi A, Kaltenbach S, Karanian M, Orbach D, Fraitag S (January 2021). "Dermatofibrosarcoma protuberans, fibrosarcomatous variant: A rare tumor in children". Pediatric Dermatology. 38 (1): 217–222. doi:10.1111/pde.14393. PMID 33010051. S2CID 222159613.
- 1 2 3 4 5 Erdem O, Wyatt AJ, Lin E, Wang X, Prieto VG (February 2012). "Dermatofibrosarcoma protuberans treated with wide local excision and followed at a cancer hospital: prognostic significance of clinicopathologic variables". The American Journal of Dermatopathology. 34 (1): 24–34. doi:10.1097/DAD.0b013e3182120671. PMID 21785324. S2CID 1402786.
- 1 2 3 4 5 Kim J, Yasuda M, Suto M, Kishi C, Motegi SI, Okamoto M, Ishikawa O (December 2019). "Unresectable local recurrence of dermatofibrosarcoma protuberans with fibrosarcomatous change treated with carbon-ion radiotherapy after neoadjuvant chemotherapy". The Journal of Dermatology. 46 (12): e457–e458. doi:10.1111/1346-8138.15056. PMID 31435947. S2CID 201274977.
- 1 2 3 4 5 Miyagawa T, Kadono T, Kimura T, Saigusa R, Yoshizaki A, Miyagaki T, Yamada D, Masui Y, Fujita H, Sato S (March 2017). "Pazopanib induced a partial response in a patient with metastatic fibrosarcomatous dermatofibrosarcoma protuberans without genetic translocations resistant to mesna, doxorubicin, ifosfamide and dacarbazine chemotherapy and gemcitabine-docetaxel chemotherapy". The Journal of Dermatology. 44 (3): e21–e22. doi:10.1111/1346-8138.13717. PMID 27988943. S2CID 7604809.
- 1 2 Tsuchihashi K, Kusaba H, Yamada Y, Okumura Y, Shimokawa H, Komoda M, Uchino K, Yoshihiro T, Tsuruta N, Hanamura F, Inadomi K, Ito M, Sagara K, Nakano M, Nio K, Arita S, Ariyama H, Kohashi K, Tominaga R, Oda Y, Akashi K, Baba E (May 2017). "Programmed death-ligand 1 expression is associated with fibrosarcomatous transformation of dermatofibrosarcoma protuberans". Molecular and Clinical Oncology. 6 (5): 665–668. doi:10.3892/mco.2017.1197. PMC 5431145. PMID 28515919.
- 1 2 3 4 5 Saiag P, Grob JJ, Lebbe C, Malvehy J, del Marmol V, Pehamberger H, Peris K, Stratigos A, Middelton M, Basholt L, Testori A, Garbe C (November 2015). "Diagnosis and treatment of dermatofibrosarcoma protuberans. European consensus-based interdisciplinary guideline". European Journal of Cancer (Oxford, England : 1990). 51 (17): 2604–8. doi:10.1016/j.ejca.2015.06.108. PMID 26189684.
- ↑ "COL1A1 collagen type I alpha 1 chain [Homo sapiens (Human)] - Gene - NCBI". Archived from the original on 2021-11-14. Retrieved 2021-12-14.
- ↑ Hiraki-Hotokebuchi Y, Yamada Y, Kohashi K, Yamamoto H, Endo M, Setsu N, Yuki K, Ito T, Iwamoto Y, Furue M, Oda Y (September 2017). "Alteration of PDGFRβ-Akt-mTOR pathway signaling in fibrosarcomatous transformation of dermatofibrosarcoma protuberans". Human Pathology. 67: 60–68. doi:10.1016/j.humpath.2017.07.001. hdl:2324/2348723. PMID 28711648.
- ↑ "EMILIN2 elastin microfibril interfacer 2 [Homo sapiens (Human)] - Gene - NCBI". Archived from the original on 2021-11-23. Retrieved 2021-12-14.
- ↑ Nakamura I, Kariya Y, Okada E, Yasuda M, Matori S, Ishikawa O, Uezato H, Takahashi K (December 2015). "A Novel Chromosomal Translocation Associated With COL1A2-PDGFB Gene Fusion in Dermatofibrosarcoma Protuberans: PDGF Expression as a New Diagnostic Tool". JAMA Dermatology. 151 (12): 1330–1337. doi:10.1001/jamadermatol.2015.2389. PMID 26332510.
- ↑ Ratan R, Patel SR (October 2016). "Chemotherapy for soft tissue sarcoma". Cancer. 122 (19): 2952–60. doi:10.1002/cncr.30191. PMID 27434055. S2CID 3609199.
- ↑ Lyu A, Wang Q (August 2018). "Dermatofibrosarcoma protuberans: A clinical analysis". Oncology Letters. 16 (2): 1855–1862. doi:10.3892/ol.2018.8802. PMC 6036409. PMID 30008876.