ERCC2



ERCC2, or XPD is a protein involved in transcription-coupled nucleotide excision repair.
The XPD (ERCC2) gene encodes for a 2.3-kb mRNA containing 22 exons and 21 introns. The XPD protein contains 760 amino acids and is a polypeptide with a size of 87kDa. Defects in this gene can result in three different disorders: the cancer-prone syndrome xeroderma pigmentosum complementation group D, photosensitive trichothiodystrophy, and Cockayne syndrome.[5]
Just like XPB, XPD is a part of human transcriptional initiation factor TFIIH and has ATP-dependent helicase activity.[6] It belongs to the RAD3/XPD subfamily of helicases.
XPD is essential for the viability of cells. Deletion of XPD in mice is lethal for developing embryos.[7]
Consequences of mutations in ERCC2
The ERCC2/XPD protein participates in nucleotide excision repair and is used in unwinding the DNA double helix after damage is initially identified. Nucleotide excison repair is a multi-step pathway that removes a wide range of different damages that distort normal base pairing. Such damages include bulky chemical adducts, ultraviolet-induced pyrimidine dimers, and several forms of oxidative damage. Mutations in the ERCC2/XPD gene can lead to various syndromes, either xeroderma pigmentosum (XP), trichothiodystrophy (TTD) or a combination of XP and TTD (XPTTD), or a combination of XP and Cockayne syndrome (XPCS).[8] TTD and CS both display features of premature aging. These features may include sensorineural deafness, retinal degeneration, white matter hypomethylation, central nervous system calcification, reduced stature, and cachexia (loss of subcutaneous fat tissue).[8][9] XPCS and TTD fibroblasts from ERCC2/XPD mutant human and mouse show evidence of defective repair of oxidative DNA damages that may underlie the segmental progeroid (premature aging) symptoms[10] (see DNA damage theory of aging).
ERCC2 and Nucleotide Excision Repair
The protein named XPD is expressed under the directions of the ERCC2 gene. The XPD protein is an indispensable part of the general transcription factor IIH (TFIIH) complex, which is a group of proteins. The two vital functions of the TFIIH complex are gene transcription and repairing damaged DNA. With the help of gene transcription, the TFIIH complex is able to control the functioning of many different genes in the body and the XPD protein acts as a stabilizer. XPB is another protein in the general transcription factor IIH (TFIIH) complex and is made from the ERCC3 gene, which works in coordination with XDP protein to commence the process of gene transcription.
Ultraviolet rays emerging from the sun, various hazardous chemicals, harmful radiations, are all known parameters for the sabotage of the DNA. A normal and healthy cell has the capability to fix the DNA damages before the problems begin due to the damaged DNA. Cells use nucleotide excision repair to fix damaged DNA. As a part of the process, the double-stranded DNA that encircles the damage is separated by the TFIIH complex. The XPD protein acts as a helicase and helps with the nucleotide excision repair process by binding to the specific regions of DNA and by unwinding the two DNA spiral strands. This exposes the damaged protein which allows the other proteins to remove the damaged section and replace the impaired area with the correct DNA.[11]
ERCC2 and xeroderma pigmentosum
Xeroderma pigmentosum (XP) is associated with the lack of DNA repair mechanism and high susceptibility of cancer. A slight insufficiency in the DNA repair mechanism may result in the development of cancer. Some cancers have been recognized with the help of the relation between the single nucleotide polymorphism and genes. The XPD protein produced by the ERCC2 gene plays an important role in the process of transcription and cell death and is also known for nucleotide excision repair pathway. Various literature studies have reviewed the correlation between polymorphisms in ERCC2 and reduced DNA repair efficiency and their influence on the development of the cancers as well as interaction with environmental exposures.
The second most common cause of xeroderma pigmentosum in the United States are due to mutations in ERCC2 gene, more than twenty-five of which have been observed in people with this disease. The xeroderma pigmentosum is caused when the ERCC2 gene prevents the TFIIH complex from repairing the damaged DNA constructively.
Consequently, all the deformity collects inside the DNA, sabotaging the repair mechanism and results in the cancerous or dead cells. Thus, the people suffering from xeroderma pigmentosum are highly sensitive to the ultraviolet rays from the sunlight due to the DNA repair problems.
So, when ultraviolet rays harm the genes, the cell grows and divides in an uncontrolled fashion and is highly prone to be cancerous. Xeroderma pigmentosum have high risk of developing cancer in skin and eyes as they are the areas mostly exposed to sun. Xeroderma pigmentosum caused by ERCC2 mutations is associated with the numerable developmental neurological malfunctioning which includes; hearing loss, poor coordination, mobility issues, lack of intellectual abilities, difficulties in talking, walking, swallowing the food and seizures.
Researchers suspect that these neurological abnormalities are due to the accumulation of DNA damage despite the brain not being exposed to ultraviolet rays. Other factors might cause the DNA damage in nerve cells as well.[12]
Interactions
ERCC2 has been shown to interact with:
Interactive pathway map
Click on genes, proteins and metabolites below to link to respective articles.[§ 1]
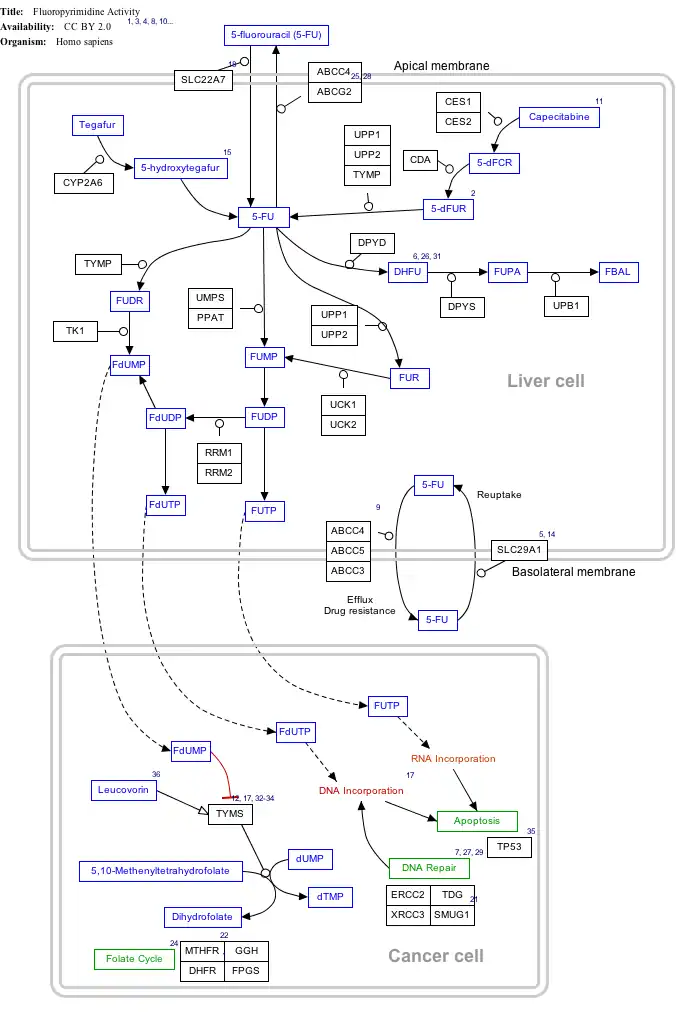
- ↑ The interactive pathway map can be edited at WikiPathways: "FluoropyrimidineActivity_WP1601".
See also
- Excision repair cross-complementing
References
- 1 2 3 GRCh38: Ensembl release 89: ENSG00000104884 - Ensembl, May 2017
- 1 2 3 GRCm38: Ensembl release 89: ENSMUSG00000030400 - Ensembl, May 2017
- ↑ "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- ↑ "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- ↑ "Entrez Gene: ERCC2 excision repair cross-complementing rodent repair deficiency, complementation group 2 (xeroderma pigmentosum D)".
- ↑ Lee TI, Young RA (2000). "Transcription of eukaryotic protein-coding genes". Annual Review of Genetics. 34: 77–137. doi:10.1146/annurev.genet.34.1.77. PMID 11092823.
- ↑ Liu, Jing. "XPD localizes in mitochondria and protects the mitochondrial genome from oxidative DNA damage". Nucleic Acids Research. 43 (11).
- 1 2 Andressoo JO, Hoeijmakers JH, Mitchell JR (2006). "Nucleotide excision repair disorders and the balance between cancer and aging". Cell Cycle. 5 (24): 2886–8. doi:10.4161/cc.5.24.3565. PMID 17172862.
- ↑ Fuss JO, Tainer JA (2011). "XPB and XPD helicases in TFIIH orchestrate DNA duplex opening and damage verification to coordinate repair with transcription and cell cycle via CAK kinase". DNA Repair (Amst.). 10 (7): 697–713. doi:10.1016/j.dnarep.2011.04.028. PMC 3234290. PMID 21571596.
- ↑ Andressoo JO, Mitchell JR, de Wit J, Hoogstraten D, Volker M, Toussaint W, Speksnijder E, Beems RB, van Steeg H, Jans J, de Zeeuw CI, Jaspers NG, Raams A, Lehmann AR, Vermeulen W, Hoeijmakers JH, van der Horst GT (2006). "An Xpd mouse model for the combined xeroderma pigmentosum/Cockayne syndrome exhibiting both cancer predisposition and segmental progeria". Cancer Cell. 10 (2): 121–32. doi:10.1016/j.ccr.2006.05.027. PMID 16904611.
- ↑ Reference, Genetics Home. "ERCC2 gene". Genetics Home Reference. Retrieved 2020-04-16.
- ↑ Benhamou, Simone; Sarasin, Alain (2002-11-01). "ERCC2/XPD gene polymorphisms and cancer risk". Mutagenesis. 17 (6): 463–469. doi:10.1093/mutage/17.6.463. ISSN 0267-8357. PMID 12435843.
- 1 2 Iyer N, Reagan MS, Wu KJ, Canagarajah B, Friedberg EC (Feb 1996). "Interactions involving the human RNA polymerase II transcription/nucleotide excision repair complex TFIIH, the nucleotide excision repair protein XPG, and Cockayne syndrome group B (CSB) protein". Biochemistry. 35 (7): 2157–67. doi:10.1021/bi9524124. PMID 8652557.
- 1 2 Drapkin R, Reardon JT, Ansari A, Huang JC, Zawel L, Ahn K, Sancar A, Reinberg D (Apr 1994). "Dual role of TFIIH in DNA excision repair and in transcription by RNA polymerase II". Nature. 368 (6473): 769–72. doi:10.1038/368769a0. PMID 8152490. S2CID 4363484.
- ↑ Rossignol M, Kolb-Cheynel I, Egly JM (Apr 1997). "Substrate specificity of the cdk-activating kinase (CAK) is altered upon association with TFIIH". The EMBO Journal. 16 (7): 1628–37. doi:10.1093/emboj/16.7.1628. PMC 1169767. PMID 9130708.
- ↑ Coin F, Marinoni JC, Rodolfo C, Fribourg S, Pedrini AM, Egly JM (Oct 1998). "Mutations in the XPD helicase gene result in XP and TTD phenotypes, preventing interaction between XPD and the p44 subunit of TFIIH". Nature Genetics. 20 (2): 184–8. doi:10.1038/2491. PMID 9771713. S2CID 28250605.
- ↑ Vermeulen W, Bergmann E, Auriol J, Rademakers S, Frit P, Appeldoorn E, Hoeijmakers JH, Egly JM (Nov 2000). "Sublimiting concentration of TFIIH transcription/DNA repair factor causes TTD-A trichothiodystrophy disorder". Nature Genetics. 26 (3): 307–13. doi:10.1038/81603. PMID 11062469. S2CID 25233797.
- ↑ Giglia-Mari G, Coin F, Ranish JA, Hoogstraten D, Theil A, Wijgers N, Jaspers NG, Raams A, Argentini M, van der Spek PJ, Botta E, Stefanini M, Egly JM, Aebersold R, Hoeijmakers JH, Vermeulen W (Jul 2004). "A new, tenth subunit of TFIIH is responsible for the DNA repair syndrome trichothiodystrophy group A". Nature Genetics. 36 (7): 714–9. doi:10.1038/ng1387. PMID 15220921.
- ↑ Marinoni JC, Roy R, Vermeulen W, Miniou P, Lutz Y, Weeda G, Seroz T, Gomez DM, Hoeijmakers JH, Egly JM (Mar 1997). "Cloning and characterization of p52, the fifth subunit of the core of the transcription/DNA repair factor TFIIH". The EMBO Journal. 16 (5): 1093–102. doi:10.1093/emboj/16.5.1093. PMC 1169708. PMID 9118947.
Further reading
- Broughton BC, Thompson AF, Harcourt SA, Vermeulen W, Hoeijmakers JH, Botta E, Stefanini M, King MD, Weber CA, Cole J (Jan 1995). "Molecular and cellular analysis of the DNA repair defect in a patient in xeroderma pigmentosum complementation group D who has the clinical features of xeroderma pigmentosum and Cockayne syndrome". American Journal of Human Genetics. 56 (1): 167–74. PMC 1801309. PMID 7825573.
- Jeang KT (1998). "Tat, Tat-associated kinase, and transcription". Journal of Biomedical Science. 5 (1): 24–7. doi:10.1007/BF02253352. PMID 9570510.
- Yankulov K, Bentley D (Jun 1998). "Transcriptional control: Tat cofactors and transcriptional elongation". Current Biology. 8 (13): R447-9. doi:10.1016/S0960-9822(98)70289-1. PMID 9651670. S2CID 15480646.
- Cleaver JE, Thompson LH, Richardson AS, States JC (1999). "A summary of mutations in the UV-sensitive disorders: xeroderma pigmentosum, Cockayne syndrome, and trichothiodystrophy". Human Mutation. 14 (1): 9–22. doi:10.1002/(SICI)1098-1004(1999)14:1<9::AID-HUMU2>3.0.CO;2-6. PMID 10447254.
- Lehmann AR (Jan 2001). "The xeroderma pigmentosum group D (XPD) gene: one gene, two functions, three diseases". Genes & Development. 15 (1): 15–23. doi:10.1101/gad.859501. PMID 11156600.
- Benhamou S, Sarasin A (Nov 2002). "ERCC2/XPD gene polymorphisms and cancer risk". Mutagenesis. 17 (6): 463–9. doi:10.1093/mutage/17.6.463. PMID 12435843.
- Clarkson SG, Wood RD (Sep 2005). "Polymorphisms in the human XPD (ERCC2) gene, DNA repair capacity and cancer susceptibility: an appraisal". DNA Repair. 4 (10): 1068–74. doi:10.1016/j.dnarep.2005.07.001. PMID 16054878.
External links
- GeneReviews/NIH/NCBI/UW entry on Xeroderma Pigmentosum
- ERCC2+Protein at the US National Library of Medicine Medical Subject Headings (MeSH)