GAPO syndrome
| GAPO syndrome | |
|---|---|
 | |
| Autosomal recessive pattern is the inheritance manner of this condition | |
| Frequency | < 1 per million |
GAPO syndrome is a rare, autosomal recessive disorder that causes severe growth retardation, and has been observed fewer than 30 times before 2011.[1] GAPO is an acronym that encompasses the predominant traits of the disorder: growth retardation, alopecia, pseudoanodontia (teeth failing to emerge from the gums), and worsening optic atrophy in some subjects. Other common symptoms include premature aging, large, prominent foreheads, and delayed bone aging. GAPO syndrome typically results in premature death around age 30-40, due to interstitial fibrosis and atherosclerosis.[2]
Signs and symptoms
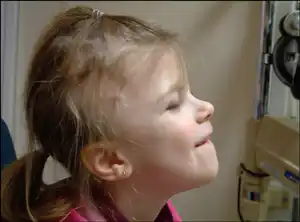
One of the principle symptoms of GAPO syndrome is growth retardation, caused by slow skeletal formation and results in individuals being below average height. Alopecia, or hair loss, is another key indication of GAPO syndrome. Their hair is typically thinly dispersed, and fragile, which often leads to baldness later in life. Similarly, tooth growth is stunted, with teeth failing to emerge form the gums or otherwise develop normally. Atrophy of the optic nerve occurs in approximately one third of individuals. This degradation leads to inhibited peripheral vision, and increased difficulty distinguishing colours.[2]
While not a defining feature, most sufferers of GAPO syndrome have coarse facial features, and abnormal structure of the middle portion of their faces, typically coupled with a large forehead.[2] Individuals with the disease tend to have depressed nose bridges, protruding ears, and abnormally thick lips, though these symptoms are not unique to this disorder.[3]
No direct correlation has been found between GAPO syndrome and mental retardation, though cases of individuals having both have been reported.[2][4]
Due to the severity of the phenotype, GAPO syndrome can be diagnosed very early on. Most cases can be diagnosed by 6 months of age, and most symptoms will be apparent by age 2.[4]
Genetics
GAPO syndrome is caused by a deletion in both copies of the ANTXR1 gene, which encodes Anthrax Toxin Receptor 1. This gene is critical for the creation of actin, and its disruption inhibits proper function of the actin network. As a result, individuals with GAPO syndrome have a buildup of extracellular matrix, and degraded cell adhesions.[3] The alteration can occur in the form of nonsense mutations or mutations which alter the splice sites, and result in alternative RNA splicing, leading to synthesis of a different or modified protein. In humans, the ANTXR1 gene is located on Chromosome 2 and has 22 exons.[5][6]
GAPO syndrome is inherited in an autosomal recessive fashion, and requires both parents to pass on the mutant genotype. Since this mutation is so rare, most confirmed cases have a history of ancestral inbreeding.[7]
Diagnosis
APO syndrome is a very rare genetic disorder characterized by growth retardation, alopecia, pseudoanodontia and progressive optic atrophy (GAPO). To date, only 30 cases have been described worldwide. Recently, gene alterations in the ANTXR1 gene have been reported to be causative of this disorder, and an autosomal recessive pattern has been observed. This gene encodes a matrix-interacting protein that works as an adhesion molecule. In this report, we describe 2 homozygous siblings diagnosed with GAPO syndrome carrying a new missense mutation. This mutation produces the substitution of a glutamine in position 137 for a leucine (c.410A>T, p.Q137L).
Management
There is currently no cure for GAPO syndrome, but some options are available to reduce the symptoms. Nearsightedness, which affects some sufferers of the disease, can be treated by corrective lenses. Unfortunately, optic atrophy as a result of degradation of the optic nerve (common with GAPO syndrome) cannot be corrected. Corticosteroids have been proposed as a treatment for optic nerve atrophy, but their effectiveness is disputed, and no steroid based treatments are currently available.[1]
History
The first incidence of GAPO syndrome was reported by Anderson and Pindborg in 1947. Another case wasn't recorded until 1978 by Fuks et al.[4]
References
- 1 2 "GAPO Syndrome". Nature's Food Patch. Natural Standard Research Collaboration. Retrieved 18 October 2015.
- 1 2 3 4 Goloni-Bertollo, Eny Maria; Ruiz, Mariangela Torreglosa; Goloni, Cristina Vendrame; Muniz, Marcos Pontes; Valério, Nelson Iguimar; Pavarino-Bertelli, Érika Cristina (2008). "GAPO syndrome: Three new Brazilian cases, additional osseous manifestations, and review of the literature". American Journal of Medical Genetics. 146A (12): 1523–1529. doi:10.1002/ajmg.a.32157. PMID 18470892. S2CID 5511228.
- 1 2 O'Neill, Marla; Sobreira, Nara; McKusick, Victor. "GAPO Syndrome". Online Mendelian Inheritance in Man. Johns Hopkins University. Archived from the original on 11 December 2015. Retrieved 14 October 2015.
- 1 2 3 Tipton, Robert; Gorlin, Robert (1984). "Growth Retardation, Alopecia, Pseudo- Anodontia, and Optic Atrophy-The GAPO Syndrome: Report of a Patient and Review of the Literature". American Journal of Medical Genetics. 19 (2): 209–216. doi:10.1002/ajmg.1320190202. PMID 6507471.
- ↑ "ANTXR1 ANTXR cell adhesion molecule 1 [Homo sapiens (human)] - Gene - NCBI".
- ↑ "ANTXR1 anthrax toxin receptor 1 [Homo sapiens (human)]". National Centre for Biotechnology Information. U.S. National Library of Medicine. Retrieved 15 October 2015.
- ↑ Bacon, W; Hall, RK; Roset, JP; Boukari, A; Tenenbaum, H; Walter, B (1999). "GAPO syndrome: a new case of this rare syndrome and a review of the relative importance of different phenotypic features in diagnosis". Journal of Craniofacial Genetics and Developmental Biology. 19 (4): 189–200. PMID 10731088.
External links
- GAPO syndrome at NIH's Office of Rare Diseases