H3K36me
H3K36me is an epigenetic modification to the DNA packaging protein Histone H3, specifically, the mono-methylation at the 36th lysine residue of the histone H3 protein.
There are diverse modifications at H3K36, such as phosphorylation, methylation, acetylation, and ubiquitylation, which have many important biological processes.[1] The methylation of H3K36 has particularly had effects in transcriptional repression, alternative splicing, dosage compensation, DNA replication and repair, DNA methylation, and the transmission of the memory of gene expression from parents to offspring during development.[1]
Nomenclature
H3K36me2 indicates dimethylation of lysine 36 on histone H3 protein subunit:[2]
| Abbr. | Meaning |
| H3 | H3 family of histones |
| K | standard abbreviation for lysine |
| 36 | position of amino acid residue
(counting from N-terminus) |
| me | methyl group |
| 1 | number of methyl groups added |
Lysine methylation
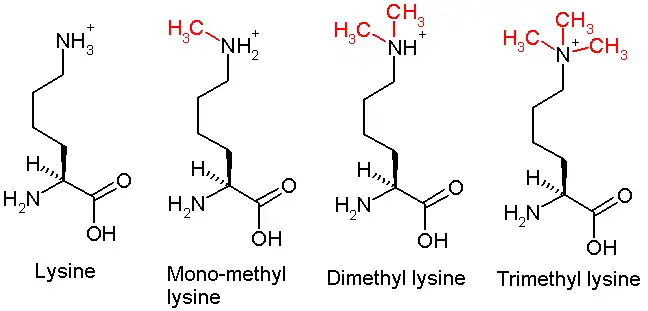
This diagram shows the progressive methylation of a lysine residue. The mono-methylation denotes the methylation present in H3K36me1.
Lysine methylation is the addition of a methyl group to the lysine of histone proteins.[3] This occurs via histone lysine methyltransferase (HMTase) that utilize S-adenosylmethionine to specifically place the methyl group on histone Lys or Arg residues.[1] So far, there have only been eight specific mammalian enzymes discovered that can methylate H3K36 in vitro and/or in vivo, all of which have identical catalytic SET domains but, different preferences for Lys36 residues in different methylation states.[1]
Histone modifications
The genomic DNA of eukaryotic cells is wrapped around special protein molecules known as histones. The complexes formed by the looping of the DNA are known as chromatin. The basic structural unit of chromatin is the nucleosome, which consists of the core octamer of histones (H2A, H2B, H3, and H4) as well as a linker histone and about 180 base pairs of DNA wrapped around it. These core histones are rich in lysine and arginine residues. The carboxyl (C) terminal end of these histones contribute to histone-histone interactions, as well as histone-DNA interactions. The amino (N) terminal charged tails are the site of the post-translational modifications, such as the one seen in H3K36me3.[4][5]
Epigenetic implications
The post-translational modification of histone tails by either histone-modifying complexes or chromatin remodeling complexes is interpreted by the cell and leads to the complex, combinatorial transcriptional output. It is thought that a histone code dictates the expression of genes by a complex interaction between the histones in a particular region.[6] The current understanding and interpretation of histones come from two large scale projects: ENCODE and the Epigenomic roadmap.[7] The purpose of the epigenomic study was to investigate epigenetic changes across the entire genome. This led to chromatin states which define genomic regions by grouping the interactions of different proteins and/or histone modifications together. Chromatin states were investigated in Drosophila cells by looking at the binding location of proteins in the genome. The use of ChIP-sequencing revealed regions in the genome characterized by different banding.[8] Different developmental stages were profiled in Drosophila as well, an emphasis was placed on histone modification relevance.[9] A look into the data obtained led to the definition of chromatin states based on histone modifications.[10] Certain modifications were mapped and enrichment was seen to localize in certain genomic regions. Five core histone modifications were found with each respective one being linked to various cell functions.
- H3K4me3-promoters
- H3K4me1- primed enhancers
- H3K36me3-gene bodies
- H3K27me3-polycomb repression
- H3K9me3-heterochromatin
The human genome was annotated with chromatin states. These annotated states can be used as new ways to annotate a genome independently of the underlying genome sequence. This independence from the DNA sequence enforces the epigenetic nature of histone modifications. Chromatin states are also useful in identifying regulatory elements that have no defined sequence, such as enhancers. This additional level of annotation allows for a deeper understanding of cell-specific gene regulation.[11]
Methods
The histone mark H3K36me can be detected in a variety of ways:
- Chromatin Immunoprecipitation Sequencing (ChIP-sequencing) measures the amount of DNA enrichment once bound to a targeted protein and immunoprecipitated. It results in good optimization and is used in vivo to reveal DNA-protein binding occurring in cells. ChIP-Seq can be used to identify and quantify various DNA fragments for different histone modifications along a genomic region.[12]
- Micrococcal Nuclease sequencing (MNase-seq) is used to investigate regions that are bound by well-positioned nucleosomes. The use of the micrococcal nuclease enzyme is employed to identify nucleosome positioning. Well-positioned nucleosomes are seen to have enrichment of sequences.[13]
- Assay for transposase accessible chromatin sequencing (ATAC-seq) is used to look into regions that are nucleosome-free (open chromatin). It uses hyperactive Tn5 transposon to highlight nucleosome localization.[14][15][16]
See also
- Histone methylation
- Histone methyltransferase
- Methyllysine
References
- 1 2 3 4 Wagner EJ, Carpenter PB (January 2012). "Understanding the language of Lys36 methylation at histone H3". Nature Reviews. Molecular Cell Biology. 13 (2): 115–26. doi:10.1038/nrm3274. PMC 3969746. PMID 22266761.
- ↑ Blakey CA, Litt MD (2015-11-30). "Chapter 2 - Histone modifications—models and mechanisms". In Huang S, Litt MD, Blakey CA (eds.). Epigenetic Gene Expression and Regulation. London: Elsevier/Academic Press. pp. 21–38. doi:10.1016/B978-0-12-799958-6.00002-0. ISBN 978-0-12-799958-6.
- ↑ Wang Z, Liu H (August 2019). "Lysine methylation regulates nervous system diseases". Neuropeptides. 76: 101929. doi:10.1016/j.npep.2019.04.004. PMID 31076097.
- ↑ Ruthenburg AJ, Li H, Patel DJ, Allis CD (December 2007). "Multivalent engagement of chromatin modifications by linked binding modules". Nature Reviews. Molecular Cell Biology. 8 (12): 983–94. doi:10.1038/nrm2298. PMC 4690530. PMID 18037899.
- ↑ Kouzarides T (February 2007). "Chromatin modifications and their function". Cell. 128 (4): 693–705. doi:10.1016/j.cell.2007.02.005. PMID 17320507.
- ↑ Jenuwein T, Allis CD (August 2001). "Translating the histone code". Science. 293 (5532): 1074–80. doi:10.1126/science.1063127. PMID 11498575.
- ↑ Birney E, Stamatoyannopoulos JA, Dutta A, Guigó R, Gingeras TR, Margulies EH, et al. (ENCODE Project Consortium) (June 2007). "Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project". Nature. 447 (7146): 799–816. Bibcode:2007Natur.447..799B. doi:10.1038/nature05874. PMC 2212820. PMID 17571346.
- ↑ Filion GJ, van Bemmel JG, Braunschweig U, Talhout W, Kind J, Ward LD, et al. (October 2010). "Systematic protein location mapping reveals five principal chromatin types in Drosophila cells". Cell. 143 (2): 212–24. doi:10.1016/j.cell.2010.09.009. PMC 3119929. PMID 20888037.
- ↑ Roy S, Ernst J, Kharchenko PV, Kheradpour P, Negre N, Eaton ML, et al. (modENCODE Consortium) (December 2010). "Identification of functional elements and regulatory circuits by Drosophila modENCODE". Science. 330 (6012): 1787–97. Bibcode:2010Sci...330.1787R. doi:10.1126/science.1198374. PMC 3192495. PMID 21177974.
- ↑ Kharchenko PV, Alekseyenko AA, Schwartz YB, Minoda A, Riddle NC, Ernst J, et al. (March 2011). "Comprehensive analysis of the chromatin landscape in Drosophila melanogaster". Nature. 471 (7339): 480–5. Bibcode:2011Natur.471..480K. doi:10.1038/nature09725. PMC 3109908. PMID 21179089.
- ↑ Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, et al. (Roadmap Epigenomics Consortium) (February 2015). "Integrative analysis of 111 reference human epigenomes". Nature. 518 (7539): 317–30. Bibcode:2015Natur.518..317.. doi:10.1038/nature14248. PMC 4530010. PMID 25693563.
- ↑ "Whole-Genome Chromatin IP Sequencing (ChIP-Seq)"(PDF). Illumina. Retrieved 23 October 2019.
- ↑ "MAINE-Seq/Mnase-Seq". illumina. Retrieved 23 October2019.
- ↑ Buenrostro JD, Wu B, Chang HY, Greenleaf WJ (January 2015). "ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide". Current Protocols in Molecular Biology. 109 (1): 21.29.1–21.29.9. doi:10.1002/0471142727.mb2129s109. PMC 4374986. PMID 25559105.
- ↑ Schep AN, Buenrostro JD, Denny SK, Schwartz K, Sherlock G, Greenleaf WJ (November 2015). "Structured nucleosome fingerprints enable high-resolution mapping of chromatin architecture within regulatory regions". Genome Research. 25 (11): 1757–70. doi:10.1101/gr.192294.115. PMC 4617971. PMID 26314830.
- ↑ Song L, Crawford GE (February 2010). "DNase-seq: a high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells". Cold Spring Harbor Protocols. 2010 (2): pdb.prot5384. doi:10.1101/pdb.prot5384. PMC 3627383. PMID 20150147.