Hereditary persistence of fetal hemoglobin
| Hereditary persistence of fetal hemoglobin | |
|---|---|
| Other names: Hereditary persistence of foetal haemoglobin | |
| Specialty | Pediatrics |
Hereditary persistence of fetal hemoglobin (HPFH) is a benign condition in which increased fetal hemoglobin (hemoglobin F, HbF) production continues well into adulthood, disregarding the normal shutoff point after which only adult-type hemoglobin should be produced.[1]
Sign and symptoms
The condition is asymptomatic, and is only noticed when screening for other hemoglobin disorders.
Sickle cell disease
In persons with sickle cell disease, high levels of fetal hemoglobin as found in a newborn or as found abnormally in persons with hereditary persistence of fetal hemoglobin, the HbF causes the sickle cell disease to be less severe. In essence the HbF inhibits polymerization of HbS. A similar mechanism occurs with persons who have sickle cell trait. Approximately 40% of the hemoglobin is in the HbS form while the rest is in normal HbA form. The HbA form interferes with HbS polymerization.[2]
Causes
HPFH can be caused by mutations in the β globin gene cluster, or the γ gene promoter region.[1] In addition HbF levels are influenced by polymorphisms in the BCL11A gene[3] and in the MYB gene enhancer.[4] In HPFH the percentage of HbF varies from 0.8-1.0% to about 30% of the total hemoglobin, but levels as high as 100% can be seen in homozygotes for delta beta thalassemia.
Diagnosis
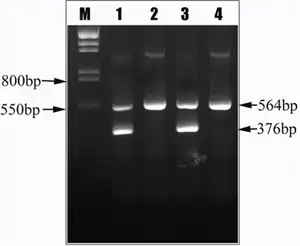
The diagnosis of Hereditary persistence of fetal hemoglobin is done via molecular genetic testing[5]
Epidemiology
About 10% of the population has an HbF level >1.0%.[1] HPFH may alleviate the severity of certain hemoglobinopathies and thalassemias, and is selected for in populations with a high prevalence of these conditions (which in turn are often selected for in areas where malaria is endemic). Thus, it has been found to affect people of African and Greek descent.[6]
References
- 1 2 3 Thein, SL; Menzel, S (May 2009). "Discovering the genetics underlying foetal haemoglobin production in adults". British Journal of Haematology. 145 (4): 455–67. doi:10.1111/j.1365-2141.2009.07650.x. PMID 19344402.
- ↑ Kumar, Vinay; Abbas, Abul K.; Fausto, Nelson; Aster, Jon (2009-05-28). Robbins and Cotran Pathologic Basis of Disease, Professional Edition: Expert Consult - Online (Robbins Pathology) (Kindle Locations 33411-33412). Elsevier Health. Kindle Edition.
- ↑ Basak, A; Sankaran, VG (March 2016). "Regulation of the fetal hemoglobin silencing factor BCL11A". Annals of the New York Academy of Sciences. 1368 (1): 25–30. Bibcode:2016NYASA1368...25B. doi:10.1111/nyas.13024. PMC 4870126. PMID 26963603.
- ↑ Stadhouders, R; Aktuna, S; Thongjuea, S; Aghajanirefah, A; Pourfarzad, F; van Ijcken, W; Lenhard, B; Rooks, H; Best, S; Menzel, S; Grosveld, F; Thein, SL; Soler, E (April 2014). "HBS1L-MYB intergenic variants modulate fetal hemoglobin via long-range MYB enhancers". The Journal of Clinical Investigation. 124 (4): 1699–710. doi:10.1172/JCI71520. PMC 3973089. PMID 24614105.
- ↑ "Hereditary persistence of fetal hemoglobin - NIH Genetic Testing Registry (GTR) - NCBI". www.ncbi.nlm.nih.gov. Archived from the original on 12 November 2022. Retrieved 9 November 2022.
- ↑ Friedman S, Schwartz E (January 1976). "Hereditary persistence of foetal haemoglobin with beta-chain synthesis in cis position (Ggamma-beta+-HPFH) in a negro family". Nature. 259 (5539): 138–40. Bibcode:1976Natur.259..138F. doi:10.1038/259138a0. PMID 1246351. S2CID 4183236.
External links
| Classification |
|---|