Hereditary inclusion body myopathy
| Hereditary inclusion body myopathy | |
|---|---|
| Other names: Hereditary inclusion body myopathy type 2 | |
Hereditary inclusion body myopathies (HIBM) are a group of rare genetic disorders which have different symptoms. Generally, they are neuromuscular disorders characterized by muscle weakness developing in young adults. Hereditary inclusion body myopathies comprise both autosomal recessive and autosomal dominant muscle disorders that have a variable expression (phenotype) in individuals, but all share similar structural features in the muscles.
HIBMs are a group of muscle wasting disorders that are uncommon in the general world population. One autosomal recessive form of HIBM is known as IBM2 or GNE myopathy, which is a common genetic disorder amongst people of Iranian Jewish descent.[1] IBM2 has also been identified in other minorities throughout the world, including those of Asian, European, and South American, and Middle Eastern descent. In Japan and other East Asian countries, this disorder is known as Distal Myopathy with Rimmed Vacuoles (DMRV).
IBM2 causes progressive muscle weakness and wasting. Muscle wasting usually starts around the age of 20 – 30 years, although young onset at 17 and old onset at 52 has been recorded. It can progress to marked disability within 10 to 15 years, confining many people with IBM2 to a wheelchair. The weakness and severity can vary from person to person. In some, weakness in the legs is noticed first. In some others, the hands are weakened more rapidly than the legs. IBM2 does not seem to affect the brain, internal organs or sensation. The quadriceps are relatively spared, and remain strong until the late stages of disease, which is the reason IBM2 is often referred to as Quadriceps Sparing Myopathy (QSM).
Signs and symptoms
Some early signs of HIBMs includes:
- Difficulty walking on heels, and difficulty running;
- Weak index finger;
- Frequent loss of balance.
- On muscle biopsy, the typical finding includes inclusion bodies, rimmed vacuoles and accumulation of aberrant proteins similar to those found in senile plaques of Alzheimer's disease (amyloid beta, hyperphosphorylated tau, amongst others)
Genetics
The different forms have different mutations and inheritance patterns. See the detailed descriptions for details
Mechanisms
The exact mechanisms of these diseases are not well understood. GNE/MNK a key enzyme in the sialic acid biosynthetic pathway, and loss-of-function mutations in GNE/MNK may lead to a lack of sialic acid, which in turn could affect sialoglycoproteins. GNE knockout mice show problems similar to people with IBM and in people with IBM dystroglycan has been found to lack sialic acid. However, the part of the dystroglycan that is important in muscle function does not seem to be affected. Another protein, neural cell adhesion molecule is under-sialyated in people with IBM, but as of 2016 it had no known role in muscle function.[2]
Diagnosis
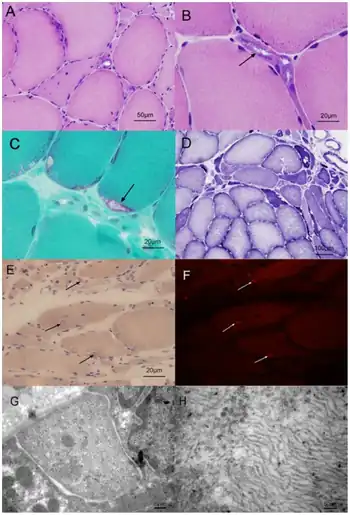
The most useful information for accurate diagnosis is the symptoms and weakness pattern. If the quadriceps are spared but the hamstrings and iliopsoas are severely affected in a person between ages of 20 - 40, it is very likely HIBM will be at the top of the differential diagnosis. The doctor may order any or all of the following tests to ascertain if a person has IBM2:
- Blood test for serum Creatine Kinase (CK or CPK);
- Nerve Conduction Study (NCS) / Electomyography (EMG);
- Muscle Biopsy;
- Magnetic Resonance Imaging (MRI) or Computer Tomography (CT) Scan to determine true sparing of quadriceps;
- Blood Test or Buccal swab for genetic testing;
Classification
Types of hereditary inclusion body myopathy:
- IBM2 is the most common form, and is an autosomal recessive form, caused by mutations in the GNE gene; this form mainly affects leg muscles, but with an unusual distribution that spares the quadriceps.[2][3][4][5] The incidence of this form is about 10 per million per year.[6] There are two forms: a quadriceps sparing myopathy (autosomal recessive form of inclusion body myopathy) and Nonaka type distal myopathy (distal myopathy with rimmed vacuoles). While occurring worldside it is most common in Jews of Persian origin. The GNE gene ecodes the rate-limiting, bifunctional enzyme of sialic acid biosynthesis, uridine diphosphate-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase. Disease occurs when both copies in the genome are non functional. The pathophysiology is still unclear. Cardiac involvement may occur.
- IBM3 is a sometimes autosomal dominant and sometimes autosomal recessive form caused by mutations in MYHC2A; it is relatively mild muscle disorder.[7][8][9]
- Inclusion body myopathy with early-onset Paget disease and frontotemporal dementia (IBMPFD), now more commonly referred to as multisystem proteinopathy (MSP), is an autosomal dominant condition caused by mutations in VCP, HNRPA2B1 or HNRNPA1; it is a multisystem degenerative disorder that can affect muscle, bone, and/or the central nervous system.[10][11][12]
The condition now called Desmin-related myofibrillar myopathy (also called myofibrillar myopathy-1) was formerly known as inclusion body myopathy 1 (IBM1).[13]
More types of HIMBs, linked to other genes, may be identified in the future.
Treatment
Treatment is palliative, not curative (as of 2009).[14]
Treatment options for lower limb weakness such as foot drop can be through the use of Ankle Foot Orthoses (AFOs) which can be designed or selected by an Orthotist based upon clinical need of the individual. Sometimes tuning of rigid AFOs can enhance knee stability.
Prognosis
A 2009 review noted that muscle weakness usually begins after age 20 and after 20–30 years, the person usually requires a wheel chair for mobility. There was no mention of increased mortality.[14]
Research
Because lack of sialic acid appears to be part of the pathology of IBM caused by GNE mutations, clinical trials with sialic acid supplements, and with a precursor of sialic acid, N-Acetylmannosamine, have been conducted, and as of 2016 further trials were planned.[2]
History
Hereditary inclusion body myopathy (IBM) constitutes a unique group of neuromuscular disorders characterized by adult-onset slowly progressive distal and proximal weakness, and a typical muscle pathology including rimmed vacuoles and filamentous inclusions. Autosomal dominant (IMB3; OMIM 605637 ) and autosomal recessive (IBM2; OMIM 600737 ) forms have been described. The autosomal recessive form, first characterized in Jews of Persian descent, is a myopathy that affects mainly leg muscles, but with an unusual distribution that spares the quadriceps, so-called quadriceps-sparing myopathy (QSM). This disorder was subsequently found in other Middle Eastern families, the gene was mapped to 9p13-p12, and in 104 affected persons from 47 Middle Eastern families the same mutation in homozygous state was found in the GNE gene.[15] Affected individuals in families of other ethnic origins were found to be compound heterozygotes for other distinct mutations in the GNE gene. From OMIM 603824. Archived 2010-03-05 at the Wayback Machine
See also
- Inclusion body myositis, a more common and non-hereditary form.
- Valosin-containing protein (VCP); mutations in VCP cause multisystem proteinopathy (MSP) which can present (among others) as a rare form of inclusion body myopathy.
References
- ↑ Pogoryelova, Oksana; González Coraspe, José Andrés; Nikolenko, Nikoletta; Lochmüller, Hanns; Roos, Andreas (December 2018). "GNE myopathy: from clinics and genetics to pathology and research strategies". Orphanet Journal of Rare Diseases. 13 (1): 70. doi:10.1186/s13023-018-0802-x. PMID 29720219.
- 1 2 3 Broccolini, Aldobrando; Mirabella, Massimiliano (April 2015). "Hereditary inclusion-body myopathies". Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 1852 (4): 644–650. doi:10.1016/j.bbadis.2014.08.007. PMID 25149037.
- ↑ "Inclusion body myopathy 2". NIH Genetic and Rare Diseases Information Center. Archived from the original on 19 September 2016. Retrieved 19 September 2016.
Alternative names include: Inclusion body myopathy, autosomal recessive; Inclusion body myopathy, quadriceps-sparing; QSM; Hereditary inclusion body myopathy; HIBM; Distal myopathy with rimmed vacuoles; DMRV; Nonaka myopathy; Rimmed vacuole myopathy; Quadriceps Sparing Myopathy; GNE myopathy
- ↑ "OMIM Entry # 605820 - Nonaka Myopathy". Online Mendelian Inheritance in Man. Archived from the original on 15 February 2020. Retrieved 19 September 2016.
- ↑ "ORPHA602: GNE myopathy". OrphaNet. Archived from the original on 19 September 2016. Retrieved 19 September 2016.
- ↑ Carrillo N, Malicdan MC, Huizing M (2018). "GNE myopathy: Etiology, diagnosis, and therapeutic challenges". Neurotherapeutics. 15 (4): 900–914. doi:10.1007/s13311-018-0671-y. PMC 6277305. PMID 30338442.
- ↑ "Inclusion body myopathy 3". NIH Genetic and Rare Diseases Information Center. Archived from the original on 19 September 2016. Retrieved 19 September 2016.
Other Names: IBM3; Myopathy with congenital joint contractures, ophthalmoplegia, and rimmed vacuoles; Inclusion body myopathy autosomal dominant; Hereditary inclusion body myopathy - joint contractures - ophthalmoplegia; Hereditary inclusion body myopathy type 3; HIBM3
- ↑ "ORPHA79091: Hereditary inclusion body myopathy-joint contractures-ophthalmoplegia syndrome". OrphaNet. Archived from the original on 19 September 2016. Retrieved 19 September 2016.
- ↑ "OMIM# 605637 - Myopathy, Proximal, and Ophthalmoplegia; MYPOP". Online Mendelian Inheritance in Man. Archived from the original on 11 May 2020. Retrieved 19 September 2016.
Myopathy With Congenital Joint Contractures, Ophthalmoplegia, And Rimmed Vacuoles Inclusion Body Myopathy 3, Autosomal Dominant, Formerly; IBM3, Formerly
- ↑ "Inclusion body myopathy with early-onset Paget disease and frontotemporal dementia". NIH - Genetic and Rare Diseases Information Center. Archived from the original on 19 September 2016. Retrieved 19 September 2016.
Other Names: IBMPFD; Inclusion body myopathy with Paget disease of bone and frontotemporal dementia; Limb-girdle muscular dystrophy with Paget disease of bone; Pagetoid amyotrophic lateral sclerosis; Pagetoid neuroskeletal syndrome
- ↑ "ORPHA52430: Inclusion body myopathy with Paget disease of bone and frontotemporal dementia". OrphaNet. Archived from the original on 19 September 2016. Retrieved 19 September 2016.
- ↑ OMIM 167320 Archived 2021-03-25 at the Wayback Machine for VCP gene; OMIM 615422 Archived 2022-01-19 at the Wayback Machine for HNRPA2B1 gene; OMIM 615424 Archived 2022-01-19 at the Wayback Machine for HNRNPA1
- ↑ "OMIM # 601419 - Myopathy, Myofibrillar 1; MFM1". Online Mendelian Inheritance in Man. Archived from the original on 10 May 2017. Retrieved 19 September 2016.
Alternative names: Myopathy, Myofibrillar, Desmin-Related. Desminopathy, Primary. Desmin-Related Myopathy; DRM. Myofibrillar Myopathy With Arrhythmogenic Right Ventricular Cardiomyopathy. Desmin-Related Myopathy With Arrhythmogenic Right Ventricular Cardiomyopathy. Arrhythmogenic Right Ventricular Dysplasia, Familial, 7, Formerly. ARVD7, Formerly. Arrhythmogenic Right Ventricular Cardiomyopathy 7, Formerly; ARVC7, Formerly. Inclusion Body Myopathy 1, Autosomal Dominant, Formerly. IBM1, Formerly. Cardiomyopathy, Dilated, 1f And Limb-Girdle Muscular Dystrophy Type 1d, Formerly. CMD1f And LGMD1d, Formerly. Cardiomyopathy, Dilated, with Conduction Defect and Muscular Dystrophy; CDCD3, formerly.
- 1 2 Huizing, Marjan; Krasnewich, Donna M. (2009-09-01). "Hereditary Inclusion Body Myopathy: A decade of progress". Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. Genetic Glycosylation Diseases. 1792 (9): 881–887. doi:10.1016/j.bbadis.2009.07.001. PMC 2748147. PMID 19596068.
- ↑ Eisenberg I, Avidan N, Potikha T, et al. (2001). "The UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase gene is mutated in recessive hereditary inclusion body myopathy". Nat. Genet. 29 (1): 83–7. doi:10.1038/ng718. PMID 11528398.
External links
- GeneReviews/NCBI/NIH/UW entry on Inclusion Body Myopathy 2 Archived 2010-04-12 at the Wayback Machine
- GeneReviews/NCBI/NIH/UW entry on Inclusion Body Myopathy with Paget Disease of Bone and/or Frontotemporal Dementia Archived 2010-05-28 at the Wayback Machine
| Classification | |
|---|---|
| External resources |
|