Multisystem proteinopathy
| Multisystem proteinopathy | |
|---|---|
| Specialty | Neurology |
| Frequency | Lua error in Module:PrevalenceData at line 5: attempt to index field 'wikibase' (a nil value). |
Multisystem proteinopathy (MSP) is a dominantly inherited, pleiotropic, degenerative disorder of humans that can affect muscle, bone, and/or the central nervous system. MSP can manifest clinically as classical amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), inclusion body myopathy (IBM), Paget's disease of bone (PDB), or as a combination of these disorders.[1] Historically, several different names have been used to describe MSP, most commonly “inclusion body myopathy with early-onset Paget disease and frontotemporal dementia (IBMPFD)” or “inclusion body myopathy with frontotemporal dementia, Paget’s disease of bone, and amyotrophic lateral sclerosis (IBMPFD/ALS).” However, IBMPFD and IBMPFD/ALS are now considered outdated classifications and are more properly referred to as MSP,[1][2] as the disease is clinically heterogeneous and its phenotypic spectrum extends beyond IBM, PDB, FTD, and ALS to include motor neuron disease, Parkinson’s disease features, and ataxia features.[3][4] Although MSP is rare, growing interest in this syndrome derives from the molecular insights the condition provides into the etiological relationship between common age-related degenerative diseases of muscle, bone, and brain.
Signs and symptoms
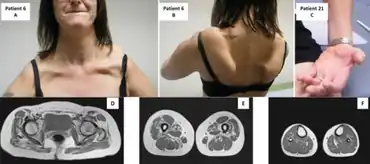
A useful operational definition of MSP is dominantly inherited degeneration that includes neurological involvement (either motor neuron disease or dementia) in combination with either distal myopathy or Pagetic bone degeneration. Most MSP patients present with weakness, and of these, approximately 65% present with motor neuron involvement.[4] Although rare, MSP can also include involvement of cardiac, hepatic, visual, auditory, sensory, and autonomic systems.[4] The histopathology of tissues affected by MSP includes ubiquitin-positive cytoplasmic inclusions of RNA-binding proteins, such as TDP-43, HNRNPA1, HNRNPA2B1, and other components of RNA granules.[3]
Genetics
MSP is a dominantly inherited and genetically heterogenous disease. The most common genetic cause of MSP is missense mutations affecting the valosin-containing protein (VCP) gene, which causes a subtype of MSP known as MSP1 (OMIM: 167320). Other pathogenic variants have been identified in HNRNPA2B1 and HNRNPA1, which cause MSP2 (OMIM: 615422) and MSP3 (OMIM: 615424) respectively. Additional genes linked to MSP include MATR3, OPTN, and p62/SQSTM1.[5]
Diagnosis
The diagnosis of this condition is based on, but not limited to the following:[6]
- Serum CK concentration
- EMG
- Elevated serum alkaline phosphatase
- Skeletal radiographs
- Genetic test
Treatment
In terms of the management of this condition, Multisystem proteinopathy we find the following:[6]
- Weight control
- Physical therapy
- Respiratory aid
- Orthopedic surgical intervention
References
- 1 2 Harrison AF, Shorter J (April 2017). "RNA-binding proteins with prion-like domains in health and disease". Biochem. J. 474 (8): 1417–1438. doi:10.1042/BCJ20160499. PMC 5639257. PMID 28389532.
- ↑ Milone M, Liewluck T (March 2019). "The unfolding spectrum of inherited distal myopathies". Muscle Nerve. 59 (3): 283–294. doi:10.1002/mus.26332. PMID 30171629. S2CID 52140733.
- 1 2 Ramaswami M, Taylor JP, Parker R (August 2013). "Altered ribostasis: RNA-protein granules in degenerative disorders". Cell. 154 (4): 727–36. doi:10.1016/j.cell.2013.07.038. PMC 3811119. PMID 23953108.
- 1 2 3 Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Stephens K, Amemiya A, Kimonis V (1993). "Inclusion Body Myopathy with Paget Disease of Bone and/or Frontotemporal Dementia". PMID 20301649.
{{cite journal}}: Cite journal requires|journal=(help) - ↑ Ralston SH, Taylor JP (May 2019). "Rare Inherited forms of Paget's Disease and Related Syndromes". Calcif. Tissue Int. 104 (5): 501–516. doi:10.1007/s00223-019-00520-5. PMC 6779132. PMID 30756140.
- 1 2 Kimonis, Virginia (1993). "Inclusion Body Myopathy with Paget Disease of Bone and/or Frontotemporal Dementia". GeneReviews®. University of Washington, Seattle. Archived from the original on 19 January 2022. Retrieved 28 January 2022.
External links
| External resources |
|---|