Inflammatory myofibroblastic tumour
| Inflammatory myofibroblastic tumour | |
|---|---|
| Other names | Epithelioid inflammatory myofibroblastic sarcoma[1] |
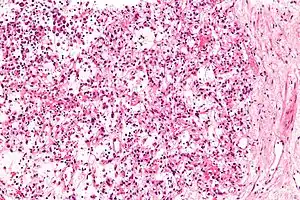 | |
| Micrograph of an inflammatory myofibroblastic tumour of the kidney. Kidney biopsy. H&E stain. | |
Inflammatory myofibroblastic tumor (IMT) is a rare neoplasm of the mesodermal cells that form the connective tissues which support virtually all of the organs and tissues of the body.[2] IMT was formerly termed inflammatory pseudotumor.[3] Currently, however, inflammatory pseudotumor designates a large and heterogeneous group of soft tissue tumors that includes inflammatory myofibroblastic tumor, plasma cell granuloma, xanthomatous pseudotumor, solitary mast cell granuloma, inflammatory fibrosarcoma,[4] pseudosarcomatous myofibroblastic proliferation, myofibroblastoma, inflammatory myofibrohistiocytic proliferation,[5] and other tumors that develop from connective tissue cells.[4] Inflammatory pseudotumour is a generic term applied to various neoplastic and non-neoplastic tissue lesions which share a common microscopic appearance consisting of spindle cells and a prominent presence of the white blood cells that populate chronic or, less commonly, acute inflamed tissues.[6][7]
Inflammatory myofibroblastic tumor was initially regarded as a benign tumor that most often developed in the lung and less commonly in almost any organ system or tissue. Over time, however, IMT cases occurred in which the tumor spread into local tissues, metastasized to distal tissues, recurred after treatment, or consisted of neoplastic cells that had pro-malignant chromosome abnormalities. Consequently, the World Health Organization, 2013, and current literature commonly describe inflammatory myofibroblastic tumor as a neoplasm with intermediate malignant potential[7] or a rarely metastasizing neoplasm.[2] In 2020, the World Health Organization reclassified IMT as a specific tumor form in the category of intermediate (rarely metastasizing) fibroblastic and myofibroblastic tumors.[8] In all events, IMT is a rare tumor with a reported incidence in 2009 of 150–200 cases/year in the United States.[9]
IMT lesions typically consist of, and are defined by, myleofibrolastic spindle cells,[7] i.e. specialized cells that are longer than wide, have a microscopic appearance that merges the appearances of fibroblasts and smooth muscle cells (see myofibroblast), occur in normal as well as tumor tissues, and in normal tissues are commonly designated fibroblasts.[10] However, the lesions in some IMF cases are dominated by sheets of epithelioid cells (which may have rounded shapes) with only a minor component of spindle cells.[11] Tumors with these characteristics are regarded as a subtype of IMT termed epithelioid inflammatory myofibroblastic sarcoma (EIMS).[3][11][12]
The tumors in IMT and EIMS consistently contain pro-inflammatory white blood cells and in most cases tumor cells that express highly abnormal oncogenic (cancer-causing) fusion proteins such as those that contain the active portion of anaplastic lymphoma kinase (ALK).[13] It is not clear whether this inflammation, the genetic abnormalities, or both contribute to the development of IMT but drugs blocking the activities of the fusion proteins made by these genetic abnormalities may be useful in treating the disease.[14]
Signs and symptoms
IMT was regarded as a tumor that occurs in children or young adults[13] and presented in the lung, mesentery, greater omentum or, less commonly, heart, liver, spleen, pancreas, colon, small intestine, spermatic cord, prostate, uterus, eye orbit, peripheral or central nervous system nerves, brain meninges, spinal cord, or other sites.[7] However, a more recent retrospective study of 92 patients accumulated by the Surveillance, Epidemiology, and End Results (SEER) program of the National Cancer Institute found the mean age of disease onset was 47.4 years with peak occurrences at 0 to 4, 36 to 40, and >50 years old; middle-aged individuals (41 to 64 years) represented 1/3 of all cases. In this study, the commonest sites of tumor occurrence were the lower limb and hip (22% of cases), upper limb and shoulder (12% of cases), and head, face, and neck (9% of cases).[7] Another recent study of 25 patients found the commonest sites of IMT were the abdomen (40% of cases) and lung/thoracic wall (32% of cases).[13] Individual IMT cases are also reported to present in the urinary bladder, anal canal, and parameningeal spaces (i.e. sites adjacent to the meninges such as the nasopharynx, middle ear, paranasal sinuses, infratemporal fossa and pterygopalatine fossa).[2] Apparently, the age and organ/tissue distribution of IMT various with the patient population examined: in general it can present in individuals of almost any age and in almost any organ or tissue site. IMT most commonly presents as a tumor localized to a single site but may be associated with distal metastases in up to 5% of all cases[7] or up to 10% of cases in which the tumor cells express an ALK fusion protein.[13] The tumors range in size from 1–25 cm (average 6.5 cm) with two-thirds being 1.5–6.5 cm.[7] In rare cases, the tumors have spontaneously regressed.[3][15]
Individuals with IMT present with a wide range of symptoms (e.g. pain, swelling, a mass, organ dysfunction, etc.) depending on the tumor location(s). Up to 1/3 of these individuals have symptoms of systemic inflammation such as fever, chills, night sweats, and weight loss.[2] Rare cases of IMT have developed in individuals with: a) organizing pneumonia; b) infection by Mycobacterium avium intracellulare or Corynebacterium equi (pneumonia-causing bacteria); Campylobacter jejuni (causes gastroenteritis); Lysinibacillus sphaericus (previously termed Bacillus sphaericus, a rare cause of lung infections[16] and sepsis);[17][18] Coxiella burneti (causes Q fever); Epstein–Barr virus (causes infectious mononucleosis and Epstein–Barr virus-associated lymphoproliferative malignant diseases); and E. coli-related occlusive phlebitis of intrahepatic veins; or c) previous abdominal surgery; trauma; ventriculoperitoneal shunt in the brain; radiation therapy; and corticosteroid usage.[19] The relationship (i.e. cause or merely association) of these disease relationships to IMT is unknown.[20]
Molecular abnormalities
The neoplastic cells in 50–60% of IMT and all cases of EIMS express an abnormal ALK protein made by a somatic recombination in the ALK gene. ALK, i.e. anaplastic lymphoma kinase (also termed protein kinase B), is produced by the ALK gene.[21] In IMT, the ALK gene has merged with a gene located at another site on the same or different chromosome to form a chimeric gene consisting of a part of the new gene and a part of the ALK gene coding for ALK's activity.[22] This chimeric gene overproduces a fusion protein with excessive ALK activity. ALK is a serine/threonine-specific protein kinase that directly or indirectly stimulates PI3K/AKT/mTOR, Ras GTPase, ERKs, Janus kinase, STAT proteins, and other cell signaling elements. Activation of these elements stimulates cell growth, proliferation, survival, and other tumor-promoting behaviors.[23][24] As an example of this chromosomal translocation, the ALK gene located on the short or "p" arm of chromosome 2 at position 23 (notated as 2p23) merges with the CLTC gene on the long, i.e. "q" arm of chromosome 17 at position 13 (notated 17q23) to form a chimeric gene notated as t(2;17)(p23;q23). This chimeric gene makes a CLTC-ALK fusion protein with uncontrolled ALK serine/threonine-specific protein kinase activity.[14][25] Other genes that fuse with AKT found in IMT include: TFG, DCTN1, EML4, TPM3,[3] TPM4,[25] ATIC[26][27] RANBP2[28] (most if not all RAMB2-ALK chimeric genes occur in the EMIS form of IMT[3]), CARS1,[11][29] and SEC31L1.[30] IMT cases may express other chimeric genes in which the active parts of ROS1 (found in 10% of IFT cases and coding for a tyrosine kinase which promotes cell growth), PDGFRB (coding for a protein that may promote the development of cancer), and NTRK (coding for a receptor tyrosine kinase that may promote the development of cancer) merge with other genes. The fusion protein products of these chimeric genes, like those of ALK fusion proteins, are overproduced, overactive, and thereby may contribute to the development of IMT.[2]
Diagnosis
Histopathologic examination of the tumors in IMT generally reveals myofibroblastic spindle cell sheets in a myxoid background (i.e. a background matrix containing gelatinous mucopolysaccharides and non-sulfated glycosaminoglycans); the matrix also contains inflammatory cells, particularly plasma cells and lymphocytes occasionally mixed with eosinophils and neutrophils. The epithelioid inflammatory myofibroblastic sarcoma subtype of IMT shows sheets of epithelioid to round cells within a myxoid (i.e. appears blue or purple compared to the normal red appearance of connective tissue when appropriately H&E stained and examined under the microscope), collagenous, or mixed myxoid-collagenous matrix, <5% spindle cells, and an inflammatory cell infiltrate that in most cases consists predominantly of neutrophils or, less often, small lymphocytes or eosinophils; plasma cells occur in only a minority of EIMS cases. The neoplastic cells in the tumors of 50% to 60% of IMT cases[2] and 100% of EIMS cases[3] express an ALK fusion protein. Other genetic abnormalities occur in these cells. Testing for the presence of the ALK fusion protein and other genetic abnormalities (see next section) can help diagnose IMT.[31]
Treatment
Many sources recommend that localized IMT be treated with total resection of all tumorous tissues.[7][13] Localized tumor recurrences may likewise be treated by total resection. There is little support for adding radiation or systemic chemotherapy to this regimen. Tumors that are not resectable, occur in inaccessible sites, are multifocal, or have metastasized are treated with aggressive therapeutic regimens.[32]
In one retrospective study, 59 patients (all <25 years old) with IMT where treated with surgery. 31 had no residual disease post-surgery; 4 of these patients had local relapses, 3 of whom were again treated surgically and 1 with surgery plus chemotherapy. Nineteen had microscopic residual disease post-surgery. Post-surgery, 6 of these patients were treated with high-dose corticosteroids; 5 with vinblastine + methotrexate chemotherapy; 3 with inhibitors of ALK; 2 with vinorelbine + low-dose cycloheximide or Ifosfamide-based chemotherapy; and 1 with cyclophosphamide + vinchristine + actinomycin D chemotherapy. Of these 19 patients, 4 had complete responses, 8 partial responses, 5 stable disease, and 2 progressive disease. Nine patients had macroscopic disease post-surgery; 5 of these patients received vincristine + methotrexate; 2 received ALK inhibitors; and 1 each received either high-dose corticosteroids or Ifosfamide-based chemotherapy. Of these 9 patients, none had complete responses; 6 had partial responses; 1 had steady disease; and 2 had progressive disease. There were no deaths among the 59 patients. The various drug regimens showed little differences in effectiveness although patients treated with ALK inhibitors trended to have longer response times.[2] Another retrospective study evaluated the response of 17 patients (aged 22–46 years; median age 32 years) with advanced disease to Adriamycin-based chemotherapy regimens, i.e. Adriamycin alone, Adriamycin + Ifosfamide, or Adriamicin + other chemotherapy drugs. No patients had a complete response, 8 patients had partial responses, 4 patients had steady disease, and 5 patients had progressive disease. Progression-free survival and overall survival times for the group were 6.6 and 21.2 months, respectively. The study also evaluated 9 patients (aged 12–31 years, median age 16) treated with methotrexate + vinblastine, methotrexate + vinorelbine, or vinblastine + vinorelbine; 2 patients attained complete responses, 3 attained partial responses, 2 had steady disease, and 2 had progressive disease; this groups' progression-free time was not reached while its overall survival time was 83.4 months. The study concluded that the Adriamycin-based and methotrexate/vinblastine/vinorelbine regimens have a high degree of activity in IMT. Due to the low numbers of patients evaluated, no conclusions could be made on which regimen(s) were most effective.[13]
In addition to the report that compared the effect of ALK-inhibitors to other therapy regimens detailed in the previous paragraph, several reports have focused primarily on small numbers of IMT patients treated with an ALK inhibitor. The European Organisation for Research and Treatment of Cancer evaluated the effect of the ALK-inhibitor, crizotinib, on 12 ALK-positive IMT adults who had persistent and/or metastatic disease following surgical and/or drug treatment: 2 patients had complete responses, 4 had partial responses, 5 had steady disease, and none had progressive disease; 9 of these patients had at least 1 year of progression-free survival but one patient died of the disease.[33] A review of previously published IMT patients of all ages found that: 1) 4 patients (3 with unifocal, 1 with multifocal disease) without prior treatment had complete responses to crizotinin; 2) 2 patients (1 with unifocal, 1 with multifocal disease) that had persistent disease after surgery and previously treated with chemotherapy had partial responses to crizotinib while 1 patient previously treated with a corticosteroid, prednisone, continued to have progressive disease on crizotinib; and 3) 6 patients with progressive disease after surgery (due to multifocal or unifocal disease in inaccessible sites) had complete responses (2 cases), partial responses (2 cases), stable disease (1 case), or progressive disease (1 case) in response to crizotinib. Two of these crizotinib-treated patients with progressive disease had near complete responses to second generation ALK inhibitors[3] Another study reviewed 29 pediatric patients (age 15 months to 17 years) who were treated with an ALK inhibitor followed by surgical removal of the tumor (5 cases), surgical tumor removal followed by an ALK inhibitor (12 cases), or an ALK inhibitor without surgery. Twelve patients had complete responses, 14 partial responses, 2 stable disease, and 2 recurrences after finishing ALK inhibitor treatment. The latter two patients obtained complete responses to retreatment with crizotinib (1 case) or a second generation ALK inhibitor, ceritinib (1 case).[14] A study of 14 pediatric patients with metastatic or inoperable ALK-positive IMT were treated with crizotinib: 5 patients obtained complete responses, 7 partial responses, and 2 stable disease. Over the study period (2–63 months), no patient developed progressive disease.[34] Numerous Medical history studies have had similar results in treated IMT with ALK inhibitors.[35][14][25][36] However, ALK inhibitors have serious side effects; in on study, crizotinib treatment was associated with pneumonia, fever of unknown cause, heart attack, sepsis, abdominal abscess, acute renal insufficiency, and the development of an abnormal EKG (i.e. QT prolongation).[33] Entrectinib, a tyrosine kinase inhibitor that is active on ROS1 and NRTK as well as AKT, has shown clinically significant activity in individual cases of patients with IMT expressing ROS1, NRTK,[37] and/or an ALK fusion proteins.[38]
See also
- Anaplastic lymphoma kinase-positive neoplasias
- ALK inhibitors
References
- ↑ "Inflammatory myofibroblastic tumor | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Retrieved 28 June 2019.
- 1 2 3 4 5 6 7 Casanova M, Brennan B, Alaggio R, Kelsey A, Orbach D, van Noesel MM, Corradini N, Minard-Colin V, Zanetti I, Bisogno G, Gallego S, Merks JH, De Salvo GL, Ferrari A (March 2020). "Inflammatory myofibroblastic tumor: The experience of the European pediatric Soft Tissue Sarcoma Study Group (EpSSG)". European Journal of Cancer. 127: 123–129. doi:10.1016/j.ejca.2019.12.021. PMID 32007712. S2CID 211012731.
- 1 2 3 4 5 6 7 Theilen TM, Soerensen J, Bochennek K, Becker M, Schwabe D, Rolle U, Klingebiel T, Lehrnbecher T (April 2018). "Crizotinib in ALK+ inflammatory myofibroblastic tumors-Current experience and future perspectives". Pediatric Blood & Cancer. 65 (4): e26920. doi:10.1002/pbc.26920. PMID 29286567. S2CID 3395900.
- 1 2 Ajani MA, Fatunla EO, Onakpoma FA, Salami AA (2020). "Inflammatory Pseudotumor: A 20-Year Single Institutional Experience". Advanced Biomedical Research. 9: 68. doi:10.4103/abr.abr_48_20. PMC 8012865. PMID 33816387.
- ↑ Savvidou OD, Sakellariou VI, Papakonstantinou O, Skarpidi E, Papagelopoulos PJ (2015). "Inflammatory myofibroblastic tumor of the thigh: presentation of a rare case and review of the literature". Case Reports in Orthopedics. 2015: 814241. doi:10.1155/2015/814241. PMC 4402203. PMID 25945274.
- ↑ Gleason BC, Hornick JL (April 2008). "Inflammatory myofibroblastic tumours: where are we now?". Journal of Clinical Pathology. 61 (4): 428–37. doi:10.1136/jcp.2007.049387. PMID 17938159.
- 1 2 3 4 5 6 7 8 Fu GX, Xu CC, Yao NF, Gu JZ, Jiang HL, Han XF (July 2019). "Inflammatory myofibroblastic tumor: A demographic, clinical and therapeutic study of 92 cases". Mathematical Biosciences and Engineering : MBE. 16 (6): 6794–6804. doi:10.3934/mbe.2019339. PMID 31698588.
- ↑ Sbaraglia M, Bellan E, Dei Tos AP (April 2021). "The 2020 WHO Classification of Soft Tissue Tumours: news and perspectives". Pathologica. 113 (2): 70–84. doi:10.32074/1591-951X-213. PMC 8167394. PMID 33179614.
- ↑ Webb TR, Slavish J, George RE, Look AT, Xue L, Jiang Q, Cui X, Rentrop WB, Morris SW (March 2009). "Anaplastic lymphoma kinase: role in cancer pathogenesis and small-molecule inhibitor development for therapy". Expert Review of Anticancer Therapy. 9 (3): 331–56. doi:10.1586/14737140.9.3.331. PMC 2780428. PMID 19275511.
- ↑ "spindle cells - Google Search". www.google.com. Retrieved 2021-11-22.
- 1 2 3 Mariño-Enríquez A, Wang WL, Roy A, Lopez-Terrada D, Lazar AJ, Fletcher CD, Coffin CM, Hornick JL (January 2011). "Epithelioid inflammatory myofibroblastic sarcoma: An aggressive intra-abdominal variant of inflammatory myofibroblastic tumor with nuclear membrane or perinuclear ALK". The American Journal of Surgical Pathology. 35 (1): 135–44. doi:10.1097/PAS.0b013e318200cfd5. PMID 21164297. S2CID 40339168.
- ↑ Telugu RB, Prabhu AJ, Kalappurayil NB, Mathai J, Gnanamuthu BR, Manipadam MT (May 2017). "Clinicopathological Study of 18 Cases of Inflammatory Myofibroblastic Tumors with Reference to ALK-1 Expression: 5-Year Experience in a Tertiary Care Center". Journal of Pathology and Translational Medicine. 51 (3): 255–263. doi:10.4132/jptm.2017.01.12. PMC 5445201. PMID 28415158.
- 1 2 3 4 5 6 Baldi GG, Brahmi M, Lo Vullo S, Cojocaru E, Mir O, Casanova M, Vincenzi B, De Pas TM, Grignani G, Pantaleo MA, Blay JY, Jones RL, Le Cesne A, Frezza AM, Gronchi A, Collini P, Dei Tos AP, Morosi C, Mariani L, Casali PG, Stacchiotti S (November 2020). "The Activity of Chemotherapy in Inflammatory Myofibroblastic Tumors: A Multicenter, European Retrospective Case Series Analysis". The Oncologist. 25 (11): e1777–e1784. doi:10.1634/theoncologist.2020-0352. PMC 7648357. PMID 32584482.
- 1 2 3 4 Craig E, Wiltsie LM, Beaupin LK, Baig A, Kozielski R, Rothstein DH, Li V, Twist CJ, Barth M (February 2021). "Anaplastic lymphoma kinase inhibitor therapy in the treatment of inflammatory myofibroblastic tumors in pediatric patients: Case reports and literature review". Journal of Pediatric Surgery. doi:10.1016/j.jpedsurg.2021.02.004. PMID 33676744. S2CID 232140059.
- ↑ Matsubayashi H, Uesaka K, Sasaki K, Shimada S, Takada K, Ishiwatari H, Ono H (October 2019). "A Pancreatic Inflammatory Myofibroblastic Tumor with Spontaneous Remission: A Case Report with a Literature Review". Diagnostics (Basel, Switzerland). 9 (4): 150. doi:10.3390/diagnostics9040150. PMC 6963339. PMID 31627359.
- ↑ Isaacson P, Jacobs PH, Mackenzie AM, Mathews AW (September 1976). "Pseudotumour of the lung caused by infection with Bacillus sphaericus". Journal of Clinical Pathology. 29 (9): 806–11. doi:10.1136/jcp.29.9.806. PMC 476182. PMID 977782.
- ↑ Castagnola E, Fioredda F, Barretta MA, Pescetto L, Garaventa A, Lanino E, Micalizzi C, Giacchino R, Dini G (June 2001). "Bacillus sphaericus bacteraemia in children with cancer: case reports and literature review". The Journal of Hospital Infection. 48 (2): 142–5. doi:10.1053/jhin.2001.0995. PMID 11428882.
- ↑ Banerjee C, Bustamante CI, Wharton R, Talley E, Wade JC (August 1988). "Bacillus infections in patients with cancer". Archives of Internal Medicine. 148 (8): 1769–74. doi:10.1001/archinte.1988.00380080059017. PMID 3401098.
- ↑ Karnak I, Senocak ME, Ciftci AO, Cağlar M, Bingöl-Koloğlu M, Tanyel FC, Büyükpamukçu N (June 2001). "Inflammatory myofibroblastic tumor in children: diagnosis and treatment". Journal of Pediatric Surgery. 36 (6): 908–12. doi:10.1053/jpsu.2001.23970. PMID 11381424.
- ↑ "Inflammatory myofibroblastic tumor | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program".
- ↑ "AKT1 AKT serine/threonine kinase 1 [Homo sapiens (human)] - Gene - NCBI". www.ncbi.nlm.nih.gov. Retrieved 2021-11-22.
- ↑ Amador C, Feldman AL (March 2021). "How I Diagnose Anaplastic Large Cell Lymphoma". American Journal of Clinical Pathology. 155 (4): 479–497. doi:10.1093/ajcp/aqab012. PMID 33686426.
- ↑ Ducray SP, Natarajan K, Garland GD, Turner SD, Egger G (July 2019). "The Transcriptional Roles of ALK Fusion Proteins in Tumorigenesis". Cancers. 11 (8): 1074. doi:10.3390/cancers11081074. PMC 6721376. PMID 31366041.
- ↑ Martorana F, Motta G, Pavone G, Motta L, Stella S, Vitale SR, Manzella L, Vigneri P (2021). "AKT Inhibitors: New Weapons in the Fight Against Breast Cancer?". Frontiers in Pharmacology. 12: 662232. doi:10.3389/fphar.2021.662232. PMC 8118639. PMID 33995085.
- 1 2 3 Cao Z, Gao Q, Fu M, Ni N, Pei Y, Ou WB (February 2019). "Anaplastic lymphoma kinase fusions: Roles in cancer and therapeutic perspectives". Oncology Letters. 17 (2): 2020–2030. doi:10.3892/ol.2018.9856. PMC 6341817. PMID 30675269.
- ↑ "ATIC 5-aminoimidazole-4-carboxamide ribonucleotide formyltransferase/IMP cyclohydrolase [Homo sapiens (Human)] – Gene – NCBI".
- ↑ Debiec-Rychter M, Marynen P, Hagemeijer A, Pauwels P (October 2003). "ALK-ATIC fusion in urinary bladder inflammatory myofibroblastic tumor". Genes, Chromosomes & Cancer. 38 (2): 187–90. doi:10.1002/gcc.10267. PMID 12939746. S2CID 40569327.
- ↑ Ma Z, Hill DA, Collins MH, Morris SW, Sumegi J, Zhou M, Zuppan C, Bridge JA (May 2003). "Fusion of ALK to the Ran-binding protein 2 (RANBP2) gene in inflammatory myofibroblastic tumor". Genes, Chromosomes & Cancer. 37 (1): 98–105. doi:10.1002/gcc.10177. PMID 12661011. S2CID 23427194.
- ↑ "SEC31A SEC31 homolog A, COPII coat complex component [Homo sapiens (Human)] – Gene – NCBI".
- ↑ Panagopoulos I, Nilsson T, Domanski HA, Isaksson M, Lindblom P, Mertens F, Mandahl N (March 2006). "Fusion of the SEC31L1 and ALK genes in an inflammatory myofibroblastic tumor". International Journal of Cancer. 118 (5): 1181–6. doi:10.1002/ijc.21490. PMID 16161041.
- ↑ Hou TC, Wu PS, Huang WY, Yang YT, Tan KT, Liu SH, Chen YJ, Chen SJ, Su YW (March 2020). "Over expression of CDK4 and MDM2 in a patient with recurrent ALK-negative mediastinal inflammatory myofibroblastic tumor: A case report". Medicine. 99 (12): e19577. doi:10.1097/MD.0000000000019577. PMC 7220190. PMID 32195970.
- ↑ "UpToDate".
- 1 2 Schöffski P, Sufliarsky J, Gelderblom H, Blay JY, Strauss SJ, Stacchiotti S, Rutkowski P, Lindner LH, Leahy MG, Italiano A, Isambert N, Debiec-Rychter M, Sciot R, Van Cann T, Marréaud S, Nzokirantevye A, Collette S, Wozniak A (June 2018). "Crizotinib in patients with advanced, inoperable inflammatory myofibroblastic tumours with and without anaplastic lymphoma kinase gene alterations (European Organisation for Research and Treatment of Cancer 90101 CREATE): a multicentre, single-drug, prospective, non-randomised phase 2 trial". The Lancet. Respiratory Medicine. 6 (6): 431–441. doi:10.1016/S2213-2600(18)30116-4. PMID 29669701. S2CID 5000248.
- ↑ Mossé YP, Voss SD, Lim MS, Rolland D, Minard CG, Fox E, Adamson P, Wilner K, Blaney SM, Weigel BJ (October 2017). "Targeting ALK With Crizotinib in Pediatric Anaplastic Large Cell Lymphoma and Inflammatory Myofibroblastic Tumor: A Children's Oncology Group Study". Journal of Clinical Oncology. 35 (28): 3215–3221. doi:10.1200/JCO.2017.73.4830. PMC 5617123. PMID 28787259.
- ↑ Mittal A, Gupta A, Dhamija E, Barwad A, Rastogi S (March 2021). "Sustained complete response on crizotinib in primary lung inflammatory myofibroblastic tumor – Case report and literature review". Monaldi Archives for Chest Disease = Archivio Monaldi per le Malattie del Torace. 91 (3). doi:10.4081/monaldi.2021.1586. PMID 33794589.
- ↑ Zhang C, Wang Z, Zhuang R, Guo X, Feng Y, Shen F, Liu W, Zhang Y, Tong H, Sun W, Liu J, Wang G, Dai C, Lu W, Zhou Y (2020). "Efficacy and Resistance of ALK Inhibitors in Two Inflammatory Myofibroblastic Tumor Patients with ALK Fusions Assessed by Whole Exome and RNA Sequencing". OncoTargets and Therapy. 13: 10335–10342. doi:10.2147/OTT.S270481. PMC 7568619. PMID 33116613.
- ↑ Ambati SR, Slotkin EK, Chow-Maneval E, Basu EM (2018). "Entrectinib in Two Pediatric Patients With Inflammatory Myofibroblastic Tumors Harboring ROS1 or ALK Gene Fusions". JCO Precision Oncology. 2 (2): 1–6. doi:10.1200/PO.18.00095. PMC 7594679. PMID 33134769.
- ↑ Bonvini P, Rossi E, Zin A, Manicone M, Vidotto R, Facchinetti A, Tombolan L, Affinita MC, Santoro L, Zamarchi R, Bisogno G (2021). "Case Report: Circulating Tumor Cells as a Response Biomarker in ALK-Positive Metastatic Inflammatory Myofibroblastic Tumor". Frontiers in Pediatrics. 9: 652583. doi:10.3389/fped.2021.652583. PMC 8116882. PMID 33996693.
External links
- ↑ DEMIR, Ömer Faruk, et al. Surgical treatment outcomes of pulmonary inflammatory myofibroblastic tumors. Annals of Thoracic Medicine, 2022, 17.1: 44.