Kataegis
In molecular biology, kataegis describes a pattern of localized hypermutations identified in some cancer genomes, in which a large number of highly patterned basepair mutations occur in a small region of DNA.[1] The mutational clusters are usually several hundred basepairs long, alternating between a long range of C→T substitutional pattern and a long range of G→A substitutional pattern. This suggests that kataegis is carried out on only one of the two template strands of DNA during replication.[1] Compared to other cancer-related mutations, such as chromothripsis, kataegis is more commonly seen; it is not an accumulative process but likely happens during one cycle of replication.[2]
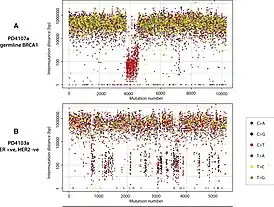
The term kataegis (καταιγίς) is derived from the ancient Greek word for "thunderstorm". It was first used by scientists at the Wellcome Trust Sanger Institute to describe their observations of breast cancer cells. In the process of mapping mutation clusters across the genome, they used a visualization tool called "rainfall plots", as shown on the picture on the right, with which they observed a clustering pattern for kataegis.[1]
Mechanism
Regions of kataegis have been shown to be colocalised with regions of somatic genome rearrangements.[1] In these regions, known as the breakpoints, basepairs are more prone to get deleted, substituted, or translocated. Most hypotheses of the kataegis involves errors during the frequent DNA repair at the breakpoints. A collection of enzymes from the DNA repair system will come in to excise the mismatch basepair. When these enzymes try to mend the mutational damage, they unwind DNA into single strands and create lesion regions that do not have a purine/pyrimidine base. Across the lesion region, the bases in the unpaired, single-stranded DNA(ssDNA) are more accessible to the modifying enzyme groups that can cause further damage in the sequence, thus forming the mutational clusters seen in kataegis.[3]
Two enzyme families are assumed to be related to kataegis. The APOBEC("apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like") enzyme family causes predominately C→T mutations, and translesional DNA synthesis (TLS) DNA polymerase causes C→G or C→T mutations.
APOBEC enzyme family (C→T mutations)
APOBEC family is a group of cytidine deaminase enzymes that plays an important role in immune system. Its major function is to induce genetic mutations in antibodies, which need a huge variety of genes in order to bind to different antigens.[5] APOBEC family can also protect against the infection of RNA retroviruses and retrotransposons.[4] In a single-strand DNA (ssDNA), APOBEC can transfer an amine group from a cytosine(C) and turn it into a uracil(U); such mutations can deaminate the viral gene and terminate the retro-transcription process that codes RNA back to DNA.[6]
As shown in Figure 1, the base mutations in kataegis regions were found to be almost exclusively cytosine to thymine in the context of a TpC dinucleotide(p denotes the phosphoribose backbone).[1] At DNA lesion sites, APOBEC enzyme can have access to long ssDNA and induce a C→U mutations. APOBEC family is processive and can continue to induce multiple mutations in a small region.[7] If this part of DNA is replicated before such mutation is repaired, the mutation gets passed on to the subclones.[3][8] The original CG pair will become a TA pair after one round of replication, hence the predominantly seen C→T mutation in kataegis.
Among the APOBEC family, APOBEC3 subfamily are responsible for protection against retroviruses such as HIV(known to be modified by APOBEC3F and APOBEC3G).[9][10] Since their original functions include editing ssDNA, they are more likely to be responsible for causing large numbers of mutations on human ssDNA. The direct link between the APOBEC deaminases and kataegistic clusters of mutations was recently obtained by expressing hyperactive deaminase in yeast cells.[7] Recent evidence has linked the over-expression of the family member APOBEC3B with multiple human cancers, highlighting its possible contribution to genomic instability and kataegis.[11]
Meanwhile, activation-induced cytidine deaminase (AID) is shown to facilitate kataegis formation in human lymphomas.[8] AID's majorly function is to diversify the genes among immune cells. Recent research shows that AID is involved in site-specific mutations in B cell tumor, while APOBEC3 subfamily causes the non-specific, cross-genomic mutations in non-B cell tumor.[8][12]
TLS DNA polymerase (C→G and C→T mutations)
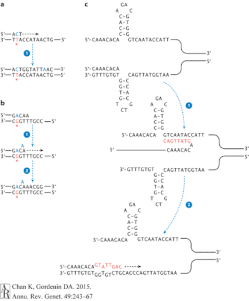
Translesional DNA synthesis (TLS) DNA polymerase family brings in the nucleotide to bridge across the abasic sites in DNA lesion. Due to the natural of the function of this enzyme, TLS DNA polymerase has a high error rates. It can slip at sequence or insert A or C base pairs into a distorted region on DNA strand; ss shown in Figure 3, TLS DNA polymerase may cause mutations in many different ways.[3]
Among the TLS DNA polymerases, Rev1 has a mechanism of inserting cytosine into lesion site that does not contain a template. Since Rev1 does not read according to Watson and Crick basepair, it can introduce any random nucleotide into the DNA sequence. In most experimental cases, Rev1 is responsible for the C→G mutation during DNA repair.[3] The effect of Rev1 can be combined with that of the APOBEC family. If the C→U mutation error is detected by its specific glycosylase, the glycosylase will cut the base pair and form an abasic site. Then TLS DNA polymerase can come in and induce C→G in this case.[12] In yeast research data, Rev1 and Rev3 can account for up 98% of basepair substitutions and 95% of UV induced mutations.[13]
Pol ζ is another kind of TLS DNA polymerase that collaborates with Rev1(mostly Rev1p) in the process of forming hypermutations in eukaryotes. Pol ζ is hypothesized to contribute to homologous allele exchanges. It can extend from DNA region distorted or bulged due to mismatches and bypass certain lesion site in DNA.[14] According to research in yeast, Pol ζ can pass different mutations with ~10% efficiency, much more often than the result from other polymerases. When Pol ζ reads pass the mutation sites, the genetic mutations remain and are passed on to the next round of replication.[13]
Clinical Significance
Kataegis is prevalently seen among breast cancer patients, and it is also exists in lung cancers, cervical, head and neck, and bladder cancers, as shown in the results from tracing APOBEC mutation signatures.[3] Understanding the mechanism of how kataegis can be useful for the future research in how cancer has developed. Due to the highly patterned mutations in kataegis, researchers can make statistical models in order to trace the loci that are prone to mutations.[3]
Research have found that kataegis could be a good prognostic indicator for breast cancer patient, that there is a life expectancy difference between patients with kataegis and those without. The specific reason was not clear. Because kataegis causes up-regulation and down-regulation of different factors, it is hypothesized that kataegis might have down-regulated the migration related gene, thus causing the tumor to be less invasive.[15]
See also
References
- 1 2 3 4 5 Nik-Zainal, Serena; Alexandrov, Ludmil B.; Wedge, David C.; Van Loo, Peter; Greenman, Christopher D.; Raine, Keiran; Jones, David; Hinton, Jonathan; Marshall, John (May 2012). "Mutational Processes Molding the Genomes of 21 Breast Cancers". Cell. 149 (5): 979–993. doi:10.1016/j.cell.2012.04.024. PMC 3414841. PMID 22608084.
- ↑ Wang, Edwin (November 2013). "Understanding genomic alterations in cancer genomes using an integrative network approach". Cancer Letters. 340 (2): 261–269. arXiv:1409.3263. doi:10.1016/j.canlet.2012.11.050. PMID 23266571. S2CID 32328448.
- 1 2 3 4 5 6 Chan, Kin; Gordenin, Dmitry A. (November 23, 2015). "Clusters of Multiple Mutations: Incidence and Molecular Mechanisms". Annual Review of Genetics. 49 (1): 243–267. doi:10.1146/annurev-genet-112414-054714. ISSN 0066-4197. PMC 4710516. PMID 26631512.
- 1 2 Prochnow, Courtney; Bransteitter, Ronda; Klein, Michael G.; Goodman, Myron F.; Chen, Xiaojiang S. (January 2017). "The APOBEC-2 crystal structure and functional implications for the deaminase AID". Nature. 445 (7126): 447–451. doi:10.1038/nature05492. ISSN 0028-0836. PMID 17187054. S2CID 4394772.
- ↑ Conticello, Silvestro G.; Thomas, Cornelia J. F.; Petersen-Mahrt, Svend K.; Neuberger, Michael S. (February 2005). "Evolution of the AID/APOBEC Family of Polynucleotide (Deoxy)cytidine Deaminases". Molecular Biology and Evolution. 22 (2): 367–377. doi:10.1093/molbev/msi026. ISSN 1537-1719. PMID 15496550.
- ↑ Smith, Harold C.; Bennett, Ryan P.; Kizilyer, Ayse; McDougall, William M.; Prohaska, Kimberly M. (May 2012). "Functions and regulation of the APOBEC family of proteins". Seminars in Cell & Developmental Biology. 23 (3): 258–268. doi:10.1016/j.semcdb.2011.10.004. PMC 4017262. PMID 22001110.
- 1 2 Lada, Artem G; Dhar, Alok; Boissy, Robert J; Hirano, Masayuki; Rubel, Aleksandr A; Rogozin, Igor B; Pavlov, Youri I (2012). "AID/APOBEC cytosine deaminase induces genome-wide kataegis". Biology Direct. 7 (1): 47. doi:10.1186/1745-6150-7-47. ISSN 1745-6150. PMC 3542020. PMID 23249472.
- 1 2 3 Casellas, Rafael; Basu, Uttiya; Yewdell, William T.; Chaudhuri, Jayanta; Robbiani, Davide F.; Di Noia, Javier M. (March 2016). "Mutations, kataegis and translocations in B cells: understanding AID promiscuous activity". Nature Reviews Immunology. 16 (3): 164–176. doi:10.1038/nri.2016.2. ISSN 1474-1733. PMC 4871114. PMID 26898111.
- ↑ Cullen, B. R. (February 1, 2006). "Role and Mechanism of Action of the APOBEC3 Family of Antiretroviral Resistance Factors". Journal of Virology. 80 (3): 1067–1076. doi:10.1128/JVI.80.3.1067-1076.2006. ISSN 0022-538X. PMC 1346961. PMID 16414984.
- ↑ Mbisa, J. L.; Bu, W.; Pathak, V. K. (May 15, 2010). "APOBEC3F and APOBEC3G Inhibit HIV-1 DNA Integration by Different Mechanisms". Journal of Virology. 84 (10): 5250–5259. doi:10.1128/JVI.02358-09. ISSN 0022-538X. PMC 2863843. PMID 20219927.
- ↑ Burns, Michael B; Temiz, Nuri A; Harris, Reuben S (September 2013). "Evidence for APOBEC3B mutagenesis in multiple human cancers". Nature Genetics. 45 (9): 977–983. doi:10.1038/ng.2701. ISSN 1061-4036. PMC 3902892. PMID 23852168.
- 1 2 Roberts, Steven A.; Gordenin, Dmitry A. (December 2014). "Hypermutation in human cancer genomes: footprints and mechanisms". Nature Reviews Cancer. 14 (12): 786–800. doi:10.1038/nrc3816. ISSN 1474-175X. PMC 4280484. PMID 25568919.
- 1 2 Lawrence, Christopher W. (June 2002). "Cellular roles of DNA polymerase ζ and Rev1 protein". DNA Repair. 1 (6): 425–435. doi:10.1016/S1568-7864(02)00038-1. PMID 12509231.
- ↑ Waters, L. S.; Minesinger, B. K.; Wiltrout, M. E.; D'Souza, S.; Woodruff, R. V.; Walker, G. C. (March 1, 2009). "Eukaryotic Translesion Polymerases and Their Roles and Regulation in DNA Damage Tolerance". Microbiology and Molecular Biology Reviews. 73 (1): 134–154. doi:10.1128/MMBR.00034-08. ISSN 1092-2172. PMC 2650891. PMID 19258535.
- ↑ D’Antonio, Matteo; Tamayo, Pablo; Mesirov, Jill P.; Frazer, Kelly A. (July 2016). "Kataegis Expression Signature in Breast Cancer Is Associated with Late Onset, Better Prognosis, and Higher HER2 Levels". Cell Reports. 16 (3): 672–683. doi:10.1016/j.celrep.2016.06.026. PMC 4972030. PMID 27373164.