Lennox–Gastaut syndrome
| Lennox–Gastaut syndrome | |
|---|---|
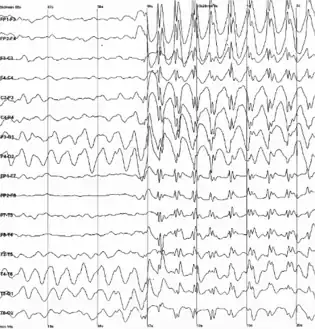 | |
| Generalized 3 Hz spike and wave discharges in a child with childhood absence epilepsy. | |
| Specialty | Neurology |
Lennox–Gastaut syndrome (LGS) is a complex, rare, and severe childhood-onset epilepsy. It is characterized by multiple and concurrent seizure types, cognitive dysfunction, and slow spike waves on electroencephalogram (EEG). Typically, it presents in children aged 3–5 years and can persist into adulthood.[1][2] It has been associated with several gene mutations, perinatal insults, congenital infections, brain tumors/malformations, and genetic disorders such as tuberous sclerosis and West syndrome. The prognosis for LGS is poor with a 5% mortality in childhood and persistent seizures into adulthood (80%–90%).[3]
LGS was named for neurologists William G. Lennox (Boston, USA) and Henri Gastaut (Marseille, France),[4] who independently described the condition. The international LGS Awareness Day is on November 1.[5]
Signs and symptoms
The symptoms vary and progress with age. The symptoms are characterized by a triad of seizures, cognitive dysfunction, and EEG findings. The triad may not fully emerge until 1–2 years after first seizure episode.
Seizures
The peak age of onset of seizures is typically between 3 and 5 years of age.[6] The mainstay symptoms is seizures that are frequent – occurring daily – and difficult to treat with antiseizure medications. An estimated 30% of patients with infantile spasms (West syndrome) have been reported to progress to LGS.[7][8]
The seizures are most commonly tonic seizures. They occur most frequently during non-REM sleep (90%). The seizures initially last only a few seconds and are activated by sleep. The presentation can be subtle. They present often as tonic eyelid opening with some changes in breathing coupled with pupillary dilation, urinary incontinence, increased heart rate, and flushing can occur.
Nonconvulsive status epilepticus occurs in about 50% of patients. The seizures can cause sudden falling often leading to injury. These "drop attacks" are typically the first manifestation of LGS. The attacks are characterized by a single, generalized monoclonic jerk that precedes tonic contraction of axial muscles.
EEG findings
Findings that strongly suggest LGS include consistent slow spike-wave (< 3 hertz [Hz]) on awake EEG. The complexes typically consist of a spike (duration < 70 milliseconds) or a sharp wave (70-200 milliseconds), followed first by a positive deep trough, then a negative wave (350-400 milliseconds). Not every wave is preceded by a spike. Bursts increase and decrease without clear onset and offset. Slow spike waves may occur during seizure or between seizures, or may occur in absence of any observable clinical changes which helps distinguish pattern from extended 3-Hz spike-wave discharges.
Ocular abnormality
Ocular abnormalities affect around 90% of children. They can present as refractive error, strabismus, cortical visual impairment, and premature retinopathy.[9]

Other
The following also are signs/symptoms experienced by individuals with this condition (although to a lesser percentage):[10]
- Frontotemporal cerebral atrophy
- Gastroesophageal reflux
- Gum enlargement
- Global developmental delay
Causes
The disease pathophysiology is mostly unknown, but some evidence implicates cortical hyperexcitability occurring at critical periods of brain development.
There are two types of LGS: idiopathic and secondary. The cause of the idiopathic subtype is unknown. Secondary LGS occurs when an identifiable underlying pathology is responsible. The most common type of LGS (70–78%) is secondary.[11] These patients tend to have a worse prognosis than those with idiopathic LGS.[12] In up to one-third of cases no cause can be found.[12]
Brain injury
Lennox–Gastaut most often occurs secondary to brain damage. The brain damage can occur from perinatal insults, encephalitis, meningitis, tumor, and brain malformation.
Genetic mutations
Other identified disorders include genetic disorders such as tuberous sclerosis and inherited deficiency of methylene tetrahydrofolate reductase. Some of these cases once thought to be of unknown cause may have definitive etiology by modern genetic testing.[12]
Progress in genome and exome sequencing is revealing that some individuals diagnosed with Lennox-Gastaut Syndrome have de novo mutations in a variety of genes, including CHD2, GABRB3, ALG13 and SCN2A.[13][14] The Epi4K study consortium (2013) observed de novo mutations in at least 15% of a study cohort of 165 patients with LGS and infantile spasms using whole exome sequencing.[15] A 2013 study found a high frequency of rare copy-number variation (CNV's) in adult patients with LGS or LGS-like epilepsy.[16]
Mutations in the IQSEC2 gene have been associated with this syndrome.[17] This gene is located on the short arm of the X chromosome (Xp11.22).
Diagnosis
The diagnosis of LGS should be suspected in children less than 8 years old with seizures of multiple types that cannot be treated with antiseizure medications. Because of high risk of irreversible brain damage in early stages of syndrome (particularly in infants and young children), early diagnosis is essential. It may take 1–2 years after first initial seizure for all criteria for diagnosis to emerge, so LGS should be considered if there are suggestive signs and symptoms without presence of complete triad.
To confirm diagnosis, awake and asleep EEG and magnetic resonance imaging (MRI) are performed. MRI is used to detect focal brain lesions.
Ruling out other diagnoses
Certain diagnoses must be ruled out before diagnosing LGS. These diagnoses are:
- Doose syndrome
- Dravet syndrome
- pseudo-Lennox Gastaut syndrome (atypical benign partial epilepsy)
LGS is more easily distinguished from Doose syndrome by seizure type after the syndrome has progressed. Doose syndrome has more myoclonic seizures and LGS has more tonic seizures. The Doose syndromes is less likely to have cognitive disabilities.
Dravet syndrome has a strong family history of epilepsy, unlike LGS. Also, many children with Dravet syndrome have seizures triggered by light.
Pseudo-Lennox–Gastaut syndrome can be distinguished from LGS because pseudo-LGS has different spike-and-wave patterns on EEG.
Treatment
There are several treatment options, including medications, surgery, and diet.
Medications
In most patients with LGS, the treatment does not end seizure recurrence. The goals of treatment are to lower frequency and severity of seizures to greatest extent possible. The appropriate treatment varies depending on individual.[18]
The treatments for LGS has evolved over the years. Various treatments have been shown to have some degree of efficacy. In 1997–1999, lamotrigine was found to be effective and approved by the Food and Drug Administration and Health Canada.[19][20][21] In 1999, topiramate trials showed that topiramate decreased seizure occurrence by more than 50%.[22][23]
Felbamate is the treatment of last resort in the event that everything else fails,[24] and was found to be superior to placebo in controlling treatment resistant partial seizures and atonic seizures.[25][26] However, it has been known to cause aplastic anemia and liver toxicity.[27]
First-line drugs
- valproate (valproic acid, sodium valproate and valproate semisodium)
Second-line drugs
Third-line drugs
Treatment of last resort
Adjuvant drugs
Are the following:
- benzodiazepines, specifically clonazepam, nitrazepam, and clobazam
- zonisamide
- cannabidiol
Surgery
In the past, LGS patients were not eligible for surgery, as the medical community thought the LGS involved the whole brain as a generalized epilepsy in all cases. Since 2010, this assumption has been challenged.[29] Two studies on LGS patients series who underwent curative surgery in Korea[30] and China,[31] showed very good results, up to seizure freedom for 80% of these patients below 5 years old, and 40% above 5 years old. Like all epilepsy curative surgeries, seizures may recur in the years following surgery, but surgery allows the child to have better brain development during the seizure free period.
There are several procedures that have shown efficacy:
- vagus nerve stimulation, which involves implantation of battery-operated generator of intermittent electrical stimuli to an electrode wrapped around left vagus nerve. Some studies have been shown it to have greater than 50% reduction in seizures reported in more than half of patients.
- corpus callosotomy, which has shown to be effective with atonic seizures. This procedure is considered in cases in which vagus nerve stimulation has failed
- transcranial direct current stimulation
- resection
Diet
A ketogenic diet is a diet that causes ketosis, a state in which there is an increased amount of ketones in the body. Adopting and maintaining rigid diet may be difficult for some families. Short-term ketogenic diet might be associated with nonsignificant decreases in frequency of parent-reported seizures in children with LGS.[32] A case series study showed 50% seizure reduction reported in almost half of children with LGS after 1 year of ketogenic diet. However, the strength of the study is challenged because it represents reports rather than scientific analysis of the clinical outcomes such as in a randomized controlled trial.[33]
Prognosis
The mortality rate ranges from 3–7% in a mean follow up period of 8.5 to 9.7 years. Death is often related to accidents.[34]
Epidemiology
LGS is seen in approximately 4% of children with epilepsy, and is more common in males than in females.[11] Usual onset is between the ages of three and five.[6] Children can have no neurological problems prior diagnosis, or have other forms of epilepsy. West syndrome is diagnosed in 20% of patients before it evolves into LGS at about 2 years old.[12]
Finland
According to a 1997 community-based retrospective study in the Helsinki metropolitan area and the province of Uusimaa, the annual incidence of Lennox–Gastaut was 2 in 100,000 (0.002%) from 1975 to 1985.[35]
United States
0.026% of all children in the Atlanta, Georgia metropolitan area were estimated to have LGS in 1997, which was defined as, "onset of multiple seizure types before age 11 years, with at least one seizure type resulting in falls, and an EEG demonstrating slow spike-wave complexes (<2.5 Hz)." The study concluded that LGS accounts for 4% of childhood epilepsies.[11]
Research
Vigabatrin was found by Feucht et al. to be an effective add-on in patients whose seizures were not satisfactorily controlled by valproate. Out of 20 children, only 1 experienced a serious side effect (dyskinesia).[36]
Zonisamide showed promise in an overview of controlled and uncontrolled trials conducted in Japan.[37] However, in a physician survey conducted December 2004, only 28% of Lennox–Gastaut and West syndrome patients improved on zonisamide.[38]
One yet to be published study from 2017 supported the use of cannabidiol.[39]A study published in the New England Journal of Medicine has shown a significant reduction of seizures in patients taking 10 and 20 mg/kg a day compared to placebo.
References
- ↑ Markand, Omkar N. (2003-12-01). "Lennox–Gastaut syndrome (childhood epileptic encephalopathy)". Journal of Clinical Neurophysiology. 20 (6): 426–441. doi:10.1097/00004691-200311000-00005. ISSN 0736-0258. PMID 14734932. S2CID 46515787.
- ↑ Archer, John S.; Warren, Aaron E. L.; Jackson, Graeme D.; Abbott, David F. (2014-01-01). "Conceptualizing Lennox–Gastaut syndrome as a secondary network epilepsy". Frontiers in Neurology. 5: 225. doi:10.3389/fneur.2014.00225. PMC 4214194. PMID 25400619.
- ↑ Asadi-Pooya, Ali A. (March 2018). "Lennox-Gastaut syndrome: a comprehensive review". Neurological Sciences. 39 (3): 403–414. doi:10.1007/s10072-017-3188-y. ISSN 1590-3478. PMID 29124439. S2CID 4243468.
- ↑ Dravet, C., & Roger, J. (1996). Henri Gastaut 1915-1995. Epilepsia, 37(4), 410–415. https://doi.org/10.1111/j.1528-1157.1996.tb00580.x Archived 2021-09-09 at the Wayback Machine
- ↑ "LGS Foundation | Lennox-Gastaut Syndrome". Archived from the original on 2021-09-08. Retrieved 2022-03-14.
- 1 2 Bourgeois, Blaise F. D.; Douglass, Laurie M.; Sankar, Raman (2014-09-01). "Lennox–Gastaut syndrome: a consensus approach to differential diagnosis" (PDF). Epilepsia. 55 Suppl 4: 4–9. doi:10.1111/epi.12567. ISSN 1528-1167. PMID 25284032. S2CID 27913979. Archived (PDF) from the original on 2022-01-20. Retrieved 2021-08-09.
- ↑ Albert P. Aldenkamp, Fritz E. Dreifuss, W. Renier, T.P.B.M. Suurmeijer, Epilepsy in Children and Adolescents. Pg. 51
- ↑ Ohtahara S, Yamatogi Y, Ohtsukd Y, Oka E, lshida T. Prognosis of West syndrome with special reference to Lennox syndrome: a developmental study. In: Wada JA, Penry JK, eds. Advunces in epileptology: The Xth Epilepsy International Symposium. New York: Raven Press, 1980: 149–54
- ↑ Kim BH, Yu YS, Kim SJ (June 2017). "Ophthalmologic Features of Lennox-Gastaut Syndrome". Korean J Ophthalmol. 31 (3): 263–267. doi:10.3341/kjo.2015.0161. PMC 5469930. PMID 28471101.
- ↑ "Lennox-Gastaut syndrome | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Archived from the original on 11 April 2021. Retrieved 8 September 2021.
- 1 2 3 Trevathan, E; Murphy, CC; Yeargin-Allsopp, M (1997). "Prevalence and descriptive epidemiology of Lennox–Gastaut syndrome among Atlanta children". Epilepsia. 38 (12): 1283–8. doi:10.1111/j.1528-1157.1997.tb00065.x. PMID 9578523. S2CID 10245400.
- 1 2 3 4 Tyagi, Satyanand; et al. (Jul–Sep 2010). "Pharmacological Management of Lennox–Gastaut Syndrome a Difficult to Treat Form of Childhood-Onset Epilepsy: An Overview" (PDF). International Journal of Pharma and Bio Sciences. 1 (3): 1–6. Archived from the original (PDF) on 2018-07-08. Retrieved 2013-09-12.
- ↑ Lund C, Brodtkorb E, Øye AM, Røsby O, Selmer KK (2014). "CHD2 mutations in Lennox–Gastaut syndrome". Epilepsy Behav. 33: 18–21. doi:10.1016/j.yebeh.2014.02.005. PMID 24614520. S2CID 140207920.
- ↑ LCapelli LP, Krepischi AC, Gurgel-Giannetti J, Mendes MF, Rodrigues T, Varela MC, Koiffmann CP, Rosenberg C (2012). "Deletion of the RMGA and CHD2 genes in a child with epilepsy and mental deficiency". Eur J Med Genet. 55 (2): 132–134. doi:10.1016/j.ejmg.2011.10.004. PMID 22178256.
- ↑ Allen AS, Berkovic SF, Cossette P, et al. (2013-09-12). "De novo mutations in the classic epileptic encephalopathies". Nature. 501 (7466): 217–221. doi:10.1038/nature12439. ISSN 0028-0836. PMC 3773011. PMID 23934111.
- ↑ Lund, Caroline; Brodtkorb, Eylert; Røsby, Oddveig; Rødningen, Olaug Kristin; Selmer, Kaja Kristine (2013-07-01). "Copy number variants in adult patients with Lennox–Gastaut syndrome features". Epilepsy Research. 105 (1–2): 110–117. doi:10.1016/j.eplepsyres.2013.01.009. ISSN 1872-6844. PMID 23415449. S2CID 6221643.
- ↑ Choi MH, Yang JO, Min JS, Lee JJ, Jun SY1, Lee YJ, Yoon JY, Jeon SJ, Byeon I, Kang JW, Kim NS (2019) A novel X-linked variant of IQSEC2 is associated with Lennox-Gastaut syndrome and mild intellectual disability in three generations of a Korean Family. Genet Test Mol Biomarkers
- ↑ Hancock, EC; Cross, JH (28 February 2013). "Treatment of Lennox–Gastaut syndrome". The Cochrane Database of Systematic Reviews (2): CD003277. doi:10.1002/14651858.CD003277.pub3. PMC 7144815. PMID 23450537.
- ↑ Motte, J; Trevathan, E; Arvidsson, JF; Barrera, MN; Mullens, EL; Manasco, P (1997). "Lamotrigine for generalized seizures associated with the Lennox–Gastaut syndrome. Lamictal Lennox–Gastaut Study Group". The New England Journal of Medicine. 337 (25): 1807–12. doi:10.1056/NEJM199712183372504. PMID 9400037.
- ↑ Epilepsy Ontario (1999). "Lamotrigine Approved in Canada for Lennox–Gastaut Syndrome". 'Sharing' News. Archived from the original on 4 February 2012. Retrieved 13 November 2005.
- ↑ Glaxo Wellcome Inc (1998). "Final Printed Labeling—Part 1". Lamictal Tablets & Chewable Dispersible Tablets (Lamotrigine) Drug Approval Page. United States Food and Drug Administration Center for Drug Evaluation and Research. Archived from the original on April 29, 2005. Retrieved 13 November 2005.
- ↑ Sachdeo, R. C.; Glauser, TA; Ritter, F; Reife, R; Lim, P; Pledger, G (1999). "A double-blind, randomized trial of topiramate in Lennox–Gastaut syndrome". Neurology. 52 (9): 1882–7. doi:10.1212/wnl.52.9.1882. PMID 10371538. S2CID 73000217. Archived from the original on 2008-12-01. Retrieved 2021-08-09.
- ↑ Alva-Moncayo, E; Ruiz-Ruiz, A (2003). "Utilidad del topiramato como terapia añadida a esquemas convencionales para el síndrome de Lennox–Gastaut" [The value of topiramate used with conventional schemes as an adjunctive therapy in the treatment of Lennox–Gastaut syndrome]. Revista de Neurología (in español). 36 (5): 453–7. doi:10.33588/rn.3605.2002014. PMID 12640599.
- ↑ "Felbatol (felbamate)". p. 3. Archived from the original on 2007-11-09. Retrieved 2007-09-19.
- ↑ The Felbamate Study Group In Lennox–Gastaut Syndrome (1993). "Efficacy of felbamate in childhood epileptic encephalopathy (Lennox-Gastaut syndrome). The Felbamate Study Group in Lennox–Gastaut Syndrome". The New England Journal of Medicine. 328 (1): 29–33. doi:10.1056/NEJM199301073280105. PMID 8347179.
- ↑ Devinsky, O; Faught, RE; Wilder, BJ; Kanner, AM; Kamin, M; Kramer, LD; Rosenberg, A (1995). "Efficacy of felbamate monotherapy in patients undergoing presurgical evaluation of partial seizures". Epilepsy Research. 20 (3): 241–6. doi:10.1016/0920-1211(94)00084-A. PMID 7796796. S2CID 21915205.
- ↑ O'neil, MG; Perdun, CS; Wilson, MB; Mcgown, ST; Patel, S (1996). "Felbamate-associated fatal acute hepatic necrosis". Neurology. 46 (5): 1457–9. doi:10.1212/wnl.46.5.1457. PMID 8628501. S2CID 46504929.
- ↑ Hakimian S, Cheng-Hakimian A, Anderson GD, Miller JW (August 2007). "Rufinamide: a new anti-epileptic medication". Expert Opin Pharmacother. 8 (12): 1931–40. doi:10.1517/14656566.8.12.1931. PMID 17696794. S2CID 19522242.
- ↑ Douglass, LM (2014). "Surgical options for patients with Lennox–Gastaut syndrome". Epilepsia. 55: 21–28. doi:10.1111/epi.12742. PMID 25284034.
- ↑ Lee, Yun Jin (2010). "Resective pediatric epilepsy surgery in Lennox–Gastaut syndrome". Pediatrics. 125 (1): e58–e66. doi:10.1542/peds.2009-0566. PMID 20008422. S2CID 4836738. Archived from the original on 2020-12-04. Retrieved 2021-08-09.
- ↑ Liu, SY (2012). "Surgical treatment of patients with Lennox–Gastaut syndrome phenotype". The Scientific World Journal. 2012: 614263. doi:10.1100/2012/614263. PMC 3353538. PMID 22629163.
- ↑ Freeman, John M. (February 2009). "Seizures, EEG events, and the ketogenic diet". Epilepsia. 50 (2): 329–330. doi:10.1111/j.1528-1167.2008.01757.x. ISSN 1528-1167. PMID 19215282.
- ↑ Cross, J. Helen (May 2012). "The ketogenic diet in the treatment of Lennox-Gastaut syndrome". Developmental Medicine and Child Neurology. 54 (5): 394–395. doi:10.1111/j.1469-8749.2012.04276.x. ISSN 1469-8749. PMID 22443688. S2CID 21003078.
- ↑ Glauser, Tracy A.; Morita, Diego A. (2002). "Introduction". Lennox–Gastaut Syndrome. eMedicine.com, Inc. Archived from the original on 19 August 2005. Retrieved 8 July 2005.
- ↑ Heiskala, H (1997). "Community-based study of Lennox–Gastaut syndrome". Epilepsia. 38 (5): 526–31. doi:10.1111/j.1528-1157.1997.tb01136.x. PMID 9184597. S2CID 23615713.
- ↑ Feucht, M; Brantner-Inthaler, S (1994). "Gamma-vinyl-GABA (vigabatrin) in the therapy of Lennox–Gastaut syndrome: an open study". Epilepsia. 35 (5): 993–8. doi:10.1111/j.1528-1157.1994.tb02544.x. PMID 7925171. S2CID 24204172.
- ↑ Yagi, K (2004). "Overview of Japanese experience-controlled and uncontrolled trials". Seizure : The Journal of the British Epilepsy Association. 13 Suppl 1: S11–5, discussion S16. doi:10.1016/j.seizure.2004.04.018. PMID 15511680.
- ↑ Yamauchi, T; Aikawa, H (2004). "Efficacy of zonisamide: our experience". Seizure : The Journal of the British Epilepsy Association. 13 Suppl 1: S41–8, discussion S49. doi:10.1016/j.seizure.2004.04.021. PMID 15511689.
- ↑ "Lennox-Gastaut Syndrome: Cannabidiol Treatment". www.neurologytimes.com. Neurology Times. 23 April 2017. Archived from the original on 8 July 2018. Retrieved 27 April 2017.
External links
| Classification | |
|---|---|
| External resources |