Panayiotopoulos syndrome
| Panayiotopoulos syndrome | |
|---|---|
| Other names: Benign childhood occipital epilepsy, Panayiotopoulos type, Early-onset benign childhood occipital epilepsy | |
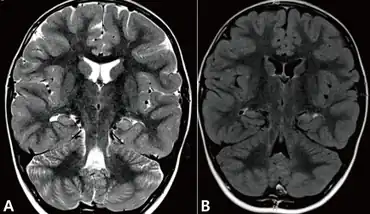 | |
| Coronal sections of brain magnetic resonance imagings taken just after the person's fourth prolonged autonomic seizure. Slightly increased signal intensity on the T2 a) and fluid-attenuated inver sion recovery image b) were observed. Structures of the left hippocampus are less obvious than the right. | |
| Specialty | Neurology |
Panayiotopoulos syndrome, also known as benign childhood occipital epilepsy, Panayiotopoulos type,[1] is a common childhood-related seizure disorder that occurs exclusively in otherwise normal children (idiopathic epilepsy) and manifests mainly with autonomic epileptic seizures and autonomic status epilepticus.[2]
It is defined as "a benign age-related focal seizure disorder occurring in early and mid-childhood. It is characterized by seizures, often prolonged, with predominantly autonomic symptoms, and by an EEG that shows shifting and/or multiple foci, often with occipital predominance."[3] It is named after Chrysostomos P. Panayiotopoulos.
Signs and symptoms
Panayiotopoulos syndrome occurs exclusively in otherwise normal children and manifests mainly with infrequent autonomic epileptic seizures and autonomic status epilepticus.[4][5][6][7] Onset of seizures is from age 1 to 14 years with 76% starting between 3–6 years. Autonomic seizures consist of episodes of disturbed autonomic function with nausea, retching and vomiting as predominant symptoms. Other autonomic manifestations include pallor (or, less often, flushing or cyanosis), mydriasis (or, less often, miosis), cardiorespiratory and thermoregulatory alterations, incontinence of urine and/or feces, hypersalivation, and modifications of intestinal motility. In approximately one fifth of the seizures the child becomes unresponsive and flaccid (syncope-like epileptic seizures or ictal syncope) before or often without convulsions. Syncope-like epileptic seizures (ictal syncope) with the child becoming "completely unresponsive and flaccid like a rag doll" occur in one fifth of the seizures.[8] More-conventional seizure symptoms often appear after the onset of autonomic manifestations. The child, who was initially fully conscious, becomes confused and unresponsive. Eyes turn to one side or gaze widely open. Only half of the seizures end with brief hemiconvulsions or generalized convulsions. Autonomic symptoms may be the only features of the seizures. None of the above symptoms alone is a prerequisite for diagnosis. Recurrent seizures may not be stereotyped. The same child may have brief or prolonged seizures and autonomic manifestations may be severe or inconspicuous. The full emetic triad (nausea, retching, vomiting) culminates in vomiting in 74% of the seizures; in others only nausea or retching occur, and in a few, none of the emetic symptoms are apparent.
Most of the seizures are prolonged and half of them last more than 30 minutes thus constituting autonomic status epilepticus, which is the more common nonconvulsive status epilepticus in normal children.[9] Characteristically, even after the most severe seizures and autonomic status epilepticus, the child is normal after a few hours of sleep, which is both diagnostic and reassuring. However, it has been recently reported that sometime after status epilepticus in children with Panayiotopoulos syndrome a. growth of the frontal and prefrontal lobes is slightly decreased and b.the scores on the neuropsychological tests is decreased.[10]
Focal onset hemiconvulsions or generalised convulsions occur in nearly half of the seizures. These are usually shorter than the preceding autonomic manifestations but in a few cases a. they may be prolonged constituting convulsive status epilepticus or b. the preceding autonomic manifestations are brief and not apparent [11]
Seizures can occur at any time but they are more common during sleep.
Cause
Panayiotopoulos syndrome is probably genetically determined, though conventional genetic influences may be less important than other mechanisms. Usually, there is no family history of similar seizures, although siblings with Panayiotopoulos syndrome or Panayiotopoulos syndrome and rolandic epilepsy or, less common, Panayiotopoulos syndrome and idiopathic childhood occipital epilepsy of Gastaut have been reported. There is a high prevalence of febrile seizures (about 17%).[12]
SCN1A mutations have been reported in a child and in 2 siblings with relatively early onset of seizures, prolonged time over which many seizures have occurred, and strong association of seizures with febrile precipitants even after the age of 5 years. However, no such mutations were found in another couple of siblings and many other cases with typical Panayiotopoulos syndrome.[13] These data indicate that SCN1A mutations when found contribute to a more severe clinical phenotype of Panayiotopoulos syndrome.
Pathophysiology
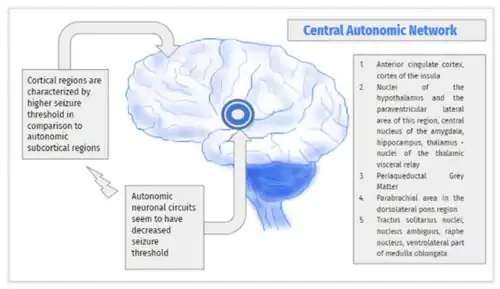
In Panayiotopoulos syndrome there is a diffuse multifocal cortical hyperexcitability, which is age (maturation)-related. This diffuse epileptogenicity may be unequally distributed, predominating in one area, which is often posterior. Epileptic discharges in Panayiotopoulos syndrome, irrespective of their location at onset, activate emetic and autonomic centers prior to any other conventional neocortical seizure manifestations. An explanation for this is that children are susceptible to autonomic disorders as illustrated by the cyclic vomiting syndrome, which is a nonepileptic condition specific to childhood.
Panayiotopoulos syndrome and all other benign childhood focal seizures, with rolandic epilepsy as their main representative, are probably linked due to a common, genetically-determined, mild, and reversible functional derangement of the brain cortical maturational process that Panayiotopoulos proposed as "benign childhood seizure susceptibility syndrome". The various EEG and seizure manifestations often follow an age- (maturation-) related localization. Panayiotopoulos syndrome is probably the early onset phenotype of the benign childhood seizure susceptibility syndrome. During a recorded autonomic seizure, there was a small increase in blood pressure (+5/4 mm Hg, systolic/diastolic), pronounced increases in heart rate (+59 bpm) and plasma concentrations of norepinephrine (+242 pg/mL), epinephrine (+175 pg/mL), and vasopressin (+22.1 pg/mL); serum glucose was also elevated (206 mg/dL).[16] The significant increase in plasma vas#pressin may explain the emetic autonomic symptoms.
Diagnosis
The most important determinant of the neurodiagnostic procedures is the state of the child at the time of first medical attendance:
(1) The child has a brief or lengthy seizure of Panayiotopoulos syndrome but fully recovers prior to arriving in the accident and emergency department or being seen by a physician. A child with the distinctive clinical features of Panayiotopoulos syndrome, particularly ictus emeticus and lengthy seizures, may not need any investigations other than EEG. However, because approximately 10% to 20% of children with similar seizures may have brain pathology, an MRI may be needed.
(2) The child with a typical lengthy seizure of Panayiotopoulos syndrome partially recovers while still in a postictal stage, tired, mildly confused, and drowsy on arrival to the accident and emergency department or when seen by a physician. The child should be kept under medical supervision until fully recovered, which usually occurs after a few hours of sleep. Then guidelines are the same as in (1) above.
(3) The child is brought to the accident and emergency department or is seen by a physician while ictal symptoms continue. This is the most difficult and challenging situation. There may be dramatic symptoms accumulating in succession, which demand rigorous and experienced evaluation. The seizure may be very dramatic, with symptoms accumulating in succession, convulsions may occur and a child who becomes unresponsive and flaccid demands rigorous and experienced evaluation. The most prominent acute disorders in the differential diagnosis include encephalitis or an encephalopathic state from causes such as infections, metabolic derangement (either inborn error or others such as hypoglycaemia), raised intracranial pressure and so forth. A history of a previous similar seizure is reassuring and may prevent further procedures.
Electroencephalography (EEG). EEG is the only investigation with abnormal results, usually showing multiple spikes in various brain locations (Figure).[17][18][19][20] There is marked variability of interictal EEG findings from normal to multifocal spikes that also change significantly in serial EEGs. Occipital spikes are common but not necessary for diagnosis. Frontal or centrotemporal spikes may be the only abnormality. Generalised discharges may happen alone or together with focal spikes. A few children have consistently normal EEG, including sleep EEG. EEG abnormalities may persist for many years after clinical remission. Conversely, spikes may appear only once in successive EEGs. Series of EEGs of the same child may present with all of the above variations from normal to very abnormal. EEG abnormalities do not appear to determine clinical manifestations, duration, severity, and frequency of seizures or prognosis.
There are now significant reports of ictal EEGs in 20 cases, which objectively document the seizures of Panayiotopoulos syndrome and their variable localisation at onset.[21] All these recorded seizures occurred while the children were asleep. The onset of the electrical ictal discharge was mainly occipital (7 cases) or frontal (7 cases) and consisted of rhythmic monomorphic decelerating theta or delta activity with small spikes. The first clinical manifestation which appeared long (1–10 minutes) after the electrical onset, usually consisted of opening of the eyes as if the children were waking from sleep. At this stage, usually the children responded, often correctly, to simple questions. On many occasions, tachycardia was the first objective sign when ||ECG|| was recorded. Vomiting was a common ictal symptom occurring at any stage of the seizures but not as the first clinical manifestation. Seizures associated with ictal vomiting did not have any particular localization or lateralization. Vomiting occurred mainly when the ictal discharges were more diffuse than localized. Sometimes only retching without vomiting occurred, and on a few occasions, vomiting did not occur. Other autonomic manifestations included mydriasis, pallor, cyanosis, tachypnea, hypersalivation, and perspiration at various stages of the ictus. Of non-autonomic manifestations, deviation of eyes to the right or left occurred before or after vomiting without any apparent EEG localisation; it was present in seizures starting from the occipital or frontal regions.
Magnetoencephalography (MEG). The multifocal nature of epileptogenicity in Panayiotopoulos syndrome has been also documented with MEG, which revealed that the main epileptogenic areas are along the parietal-occipital, the calcarine, or the central (rolandic) sulci. Patients with frontal spikes were significantly older than patients with spikes on rolandic, parieto-occipital, or calcarine sulci. Follow-up MEG demonstrated shifting localization or disappearance of MEG spikes.[22][23]
Illustrative cases
In a typical presentation of Panayiotopoulos syndrome, the child looks pale, vomits, and is fully conscious, able to speak, and understand but complains of “feeling sick.” Two thirds of the seizures start in sleep; the child may wake up with similar complaints while still conscious or else may be found vomiting, conscious, confused, or unresponsive.
Case 1. A girl had 2 seizures in sleep at 6 years of age. In the first fit she was found vomiting vigorously, eyes turned to one side, pale, and unresponsive. Her condition remained unchanged for 3 hours before she developed generalized tonic-clonic convulsions. She gradually improved, and by the next morning was normal. The second seizure occurred 4 months later. She awoke and told her mother that she wanted to vomit, and then vomited. Within minutes her eyes turned to the right. Her mother, who was on her left, asked, "Where am I?" "There, there," the child replied, indicating to the right. Ten minutes later she closed her eyes and became unresponsive. Generalized convulsions occurred 1 hour from onset. Thereafter she recovered quickly. Her EEGs showed occipital paroxysms, but this normalized by the age of 10 years. The patient had in childhood infrequent vasovagal syncopes and/or syncope-like epileptic seizures. At last communication with her, she was 29 years of age and following a successful professional career.
Case 2. This case illustrates autonomic status epilepticus with behavioral disturbances that would be difficult to attribute to seizure activity before the motor focal ictal events. A 6-year-old normal boy had a seizure at 4 years of age while traveling on a train with his parents who vividly described the event: “He was happily playing and asking questions when he started complaining that he was feeling sick, became very pale, and quiet. He did not want to drink or eat. Gradually, he was getting more and more pale, kept complaining that he felt sick, and became restless and frightened. Ten minutes from the onset, his head and eyes slowly turned to the left. The eyes were opened but fixed to the left upper corner. We called his name but he was unresponsive. He had completely gone. We tried to move his head but this was fixed to the left. There were no convulsions. This lasted for another 15 minutes, when his head and eyes returned to normal and he looked better, although he was droopy and really not there. At this stage he vomited once. In the ambulance, approximately 35 minutes from the onset, he was still not aware of what was going on, although he was able to answer simple questions with yes or no. In the hospital he slept for three quarters of an hour and gradually came around, but it took him another half to an hour before he became normal again”. EEG showed occipital paroxysms and MRI was normal. A similar prolonged episode, preceded by behavioral changes, occurred 8 months later at school. He received no medication. Since then he has been well.
Case 3. This case illustrates autonomic status epilepticus with frequent vomiting witnessed from onset. An 8-year-old boy of mixed race had 2 prolonged seizures at the age of 5 years. The first seizure occurred during a brief nap. He woke up and walked with “shaking feet” to his mother, complaining that he felt sick. Within 2 to 3 minutes, his eyes and subsequently his head turned to the extreme right. His mother recalls: “I asked him to look at me, and he would not. If I moved his head to the front, it would go back to the right. Within a minute he vomited, and his eyes started blinking, and there were also tiny jerks of his body, legs, and arms that lasted for a minute. He became unresponsive to anything I said to him. He then was rigid, and he went to a deep sleep like in a coma. In the hospital he continued to be in this unconscious state, ever so often just getting up to be sick, and straight back down again. He did not start to regain consciousness or be aware of people around until about 3 hours later. He was well the next morning and discharged home.” The second seizure occurred 6 months later on a ferryboat trip: “He told me that he felt sick, and on his way to the toilet his eyes and head turned to the right and he was talking out of context, and then he was sick. I thought he was having another fit. He was still able to converse with me in and out of sleep. He did not become unconscious, but he was continuously sick for several hours. By the time we arrived in a hospital 3 hours later, he was improving; he just seemed tired. The doctors told us that this was due to dehydration, for which treatment was provided. He was normal the next morning”. Awake EEG 1 month after the first seizure showed only 1 left-sided occipital and a possible frontal midline spike. A second EEG at 8 years of age showed infrequent central, frontal, and midline spikes during sleep.
Case 4. This case illustrates pure autonomic status epilepticus with EEG midline spikes and subsequent rolandic seizures with centrotemporal spikes. A 9-year-old boy returned from school one day looking tired and pale. Five minutes later, he complained of headache and became agitated and paler. Within 5 minutes, he started banging his head on the wall and soon became unresponsive and floppy “like a rag doll,” as well as incontinent of urine and feces with his eyes widely open and pupils markedly dilated. At this stage, he vomited vigorously. This condition continued on his way to the hospital where he arrived by ambulance half an hour from onset. Three hours later, he was still confused, partly unresponsive, pale, and quiet, and he vomited again. Recovery started 4 hours from onset. He did not convulse at any stage. He was apyrexial, and other autonomic functions were normal. He slept and was entirely normal the next morning, discharged home with the diagnosis “epileptic seizure? probably atypical migraine.” EEG had midline spikes at central midline electrode. On follow-up exam 1 year later, he had 2 typical rolandic seizures, and EEG showed centrotemporal spikes. At last follow-up at 11 years of age, he was well with no further seizures.
Case 5. This case involved seizures manifesting mainly with syncope-like epileptic seizures without emesis. A 7-year-old boy had from 5 years of age approximately 12 episodes of collapse at school. All episodes were stereotyped but of variable duration from 2 to 35 minutes. While standing or sitting, he slumped forwards and fell on his desk or the floor and became unresponsive as if in “deep sleep.” There were no convulsions or other discernible ictal or postictal symptoms. Four EEGs consistently showed frequent multifocal spikes predominating in the frontal regions.
Case 6. This case also illustrates the features of syncope-like epileptic seizures together with other variable autonomic symptoms (emesis, respiratory abnormalities, pallor, mydriasis) in Panayiotopoulos syndrome. A 5-year-old boy at age 13 months woke up vomiting profusely and then, while he was still in bed, became unresponsive and floppy with shallow breathing for 20 minutes. Later the same night, he woke up, vomited, and then collapsed in the bath. He remained flaccid and unresponsive for 1 hour, and his mother, described him as “flat” and pale with dilated nonreactive pupils. At age 20 months, he collapsed on the floor pale, unresponsive, and flaccid for approximately 10 minutes. On another occasion, he was found in bed unresponsive, floppy, and pale for 5 minutes. The last seizure occurred at age 28 months in the nursery. He fell on the floor and remained unresponsive and flaccid for 20 minutes and then he rapidly recovered. EEGs consistently showed multifocal spikes in various brain locations. Cardiologic assessment was normal.[24]
Case 7. This case demonstrates that Panayiotopoulos syndrome can also occur with consistently normal interictal EEGs.[25] At the age of 2 years, a girl had an autonomic status epilepticus during sleep. This was characterized by pallor, progressive impairment of consciousness, and vomiting that lasted 45 minutes. A second episode occurred after 11 months, during sleep, and consisted of impairment of consciousness, hypotonia, deviation of the eyes to the right, hypersalivation, and right-sided clonic convulsions. It was terminated after 45 minutes with rectal diazepam. Treatment with carbamazepine was initiated. After 6 months, she had a third episode similar to the previous ones, but shorter. At the age of 4 years 9 months, during an ambulatory EEG, she had another autonomic seizure with marked ictal EEG abnormalities, but again, the interictal did not show any spikes. Carbamazepine was replaced with phenobarbital. All 12 interictal EEGs during the active seizure period, 6 of them during sleep, were normal. At last follow-up at the age of 16 years, she was well, a good student, unmedicated, and seizure-free for 11 years.
Case 8. This case illustrates that children with Panayiotopoulos syndrome may be misdiagnosed and treated for encephalitis. This boy had a first seizure at age 4 years and 2 months. Whilst sleeping in his mother's lap, he suddenly vomited. Then his eyes stared into the space upwards, his head deviated to the right, his face turned green and he became incontinent of urine and faeces. The seizure lasted for 15 min and there were no convulsions. Abdominal ultrasound performed because of the vomiting was normal. A second seizure occurred 16 months later at age 5 years and 6 months. At around 10 in the morning he walked into lounge looking pale and irritable. He fell to the floor and developed writhing movements, shaking arms and legs, hypersalivation and incontinence of urine. Convulsions stopped 15 min later with rectal diazepam. He recovered but remained very sleepy. He was febrile ~38.5 °C. He was treated in a major teaching hospital with triple therapy for suspected encephalitis but in the third day after admission this was stopped and he was discharged home. Brain CT scan, EEG and CSF were normal. Subsequent EEGs showed infrequent occipital and frontal spikes. At follow-up aged 7, he was normal and had experienced no further seizures.[26]
Classification and nomenclature
Panayiotopoulos syndrome is now the formally approved nomenclature for this syndrome in the new International League against Epilepsy report on classification,[27] which abandoned a number of previously used descriptive terms such as early onset benign childhood epilepsy with occipital paroxysms, early onset benign childhood occipital epilepsy, nocturnal childhood occipital epilepsy. The reason for this is that these descriptive terms were criticized as incorrect because in Panayiotopoulos syndrome:
- (1) Onset of seizures is mainly with autonomic symptoms, which are not occipital lobe manifestations.
- (2) Of occipital symptoms, only deviation of the eyes may originate from the occipital regions, but this rarely occurs at onset. Visual symptoms are exceptional and not consistent in recurrent seizures.
- (3) Interictal occipital spikes may never occur.
- (4) Magnetoencephalography may show equivalent current dipoles clustering in the frontal areas.
- (5) Ictal EEG has documented variable onset from the posterior or anterior regions
“An autonomic seizure is an epileptic seizure characterized by altered autonomic function of any type at seizure onset or in which manifestations consistent with altered autonomic function are prominent (quantitatively dominant or clinically important) even if not present at seizure onset. The altered autonomic function may be objective or subjective or both.”[28]
“Autonomic status epilepticus is an autonomic seizure which lasts for more than 30 minutes, or a series of such seizures over a 30 minute period without full recovery between seizures.”[29]
Differential diagnosis
The distinctive clinical features particularly lengthy seizures and ictus emeticus means that the diagnosis of Panayiotopoulos syndrome is easy. However, these are frequently mistaken as nonepileptic conditions such as acute encephalitis, syncope, migraine, cyclic vomiting syndrome, motion sickness, sleep disorder, or gastroenteritis.[30] The consequence is avoidable misdiagnosis, high morbidity, and costly mismanagement. Autonomic seizures and autonomic status epilepticus as occur in Panayiotopoulos syndrome have not been described in other epileptic syndromes in that sequence though 10–20 per cent of children with the same seizure semiology may have cerebral pathology. The major problem is to recognize emetic and other autonomic manifestations as seizure events and not to dismiss them or erroneously to consider them as unrelated to the ictus and a feature of encephalitis, migraine, syncope or gastro-enteritis.
Management
Continuous prophylactic antiepileptic drug (AED) treatment may not be needed particularly for children with only 1-2 or brief seizures. This is probably best reserved for children whose seizures are unusually frequent, prolonged, distressing, or otherwise significantly interfering with the child's life. There is no evidence of superiority of monotherapy with any particular common AED.[31][32]
Autonomic status epilepticus in the acute stage needs thorough evaluation for proper diagnosis and assessment of the neurologic/autonomic state of the child. "Rescue" benzodiazepines are commonly used to terminate it. Aggressive treatment should be avoided because of the risk of iatrogenic complications, including cardiovascular arrest. There is some concern that intravenous lorazepam and/or diazepam may precipitate cardiovascular arrest.[33] Early parental treatment is more effective than late emergency treatment. Buccal midazolam is probably the first choice medication for out of hospital termination of autonomic status epilepticus which should be administered as soon as the child shows evidence of onset of its habitual autonomic seizures.
Parental education about Panayiotopoulos syndrome is the cornerstone of correct management. The traumatizing, sometimes long-lasting effect on parents is significant particularly because autonomic seizures may last for many hours compounded by physicians’ uncertainty regarding diagnosis, management, and prognosis.[34]
Prognosis
Panayiotopoulos syndrome is remarkably benign in terms of its evolution.[35][36][37][38][39] The risk of developing epilepsy in adult life is probably no more than of the general population. Most patients have one or 2-5 seizures. Only a third of patients may have more than 5 seizures, and these may be frequent, but outcome is again favorable. However, one fifth of patients may develop other types of infrequent, usually rolandic seizures during childhood and early teens. These are also age-related and remit before the age of 16 years. Atypical evolutions with absences and drop attacks are exceptional. Children with pre-existing neurobehavioral disorders tend to be pharmacoresistant and have frequent seizures though these also remit with age. Formal neuropsychological assessment of children with Panayiotopoulos syndrome showed that these children have normal IQ and they are not on any significant risk of developing cognitive and behavioural aberrations, which when they occur they are usually mild and reversible.[40] Prognosis of cognitive function is good even for patients with atypical evolutions.[41] However, though Panayiotopoulos syndrome is benign in terms of its evolution, autonomic seizures are potentially life-threatening in the rare context of cardiorespiratory arrest.[42]
Epidemiology
Panayiotopoulos syndrome probably affects 13% of children aged 3 to 6 years who have had 1 or more afebrile seizures and 6% of such children in the 1- to 15-year age group.[43][44] All races and both sexes are affected.
History
Chrysostomos (Tomis) P. Panayiotopoulos described this syndrome and autonomic status epilepticus particular to childhood through a 30-year prospective study that started in Greece in 1975.[45] Initial publications included patients with EEG occipital paroxysms or occipital spikes that attracted the main attention, but later it became apparent that the same clinical manifestations, and mainly ictal vomiting, could occur in children with EEG extraoccipital spikes or normal EEG.
In Panayiotopoulos’ original study, ictal vomiting occurred in only 24 children out of 900 patients of all ages with epileptic seizures.[46] Twenty-one were otherwise normal children (idiopathic cases constituting what is now considered Panayiotopoulos syndrome), and 3 had symptomatic epilepsies. Half of the seizures were lengthy, lasting for hours (autonomic status epilepticus). The EEG of the 21 idiopathic cases showed great variations: 12 had occipital paroxysms or spikes alone or with extraoccipital spikes; 2 had central spikes and giantsomatosensory evoked spikes; 2 had midline spikes; 1 had frontal spikes; 1 had brief generalized discharges; and 3 had consistently normal EEG. Subsequent attention was focused on the predominant group with occipital spikes, which was established as "early onset benign childhood epilepsy with occipital paroxysms". The other group of 9 children with extraoccipital spikes or normal EEGs was reevaluated much later; their clinical manifestations and outcome were similar to those patients with occipital spikes. Based on these results, it has been concluded that these 21 children, despite different EEG manifestations, suffered from the same disease, which is now designated as Panayiotopoulos syndrome to incorporate all cases irrespective of EEG localizations.
However, there was initial scepticism and resistance to these findings, including from influential epileptologists because as explained by Ferrie and Livingston:[47]"(a) ictal vomiting had been considered as extremely rare and hitherto had been mainly described in neurosurgical series of adult patients. In children it was generally not considered as having an epileptic origin; (b) autonomic status epilepticus was not recognised as a diagnostic entity; the proposition that it might be a common occurrence in a benign seizure disorder challenged orthodox concepts of status epilepticus; (c) it implied that paediatricians had been failing to diagnose significant numbers of children with epilepsy, instead erroneously labeling then as having diverse non-epileptic disorders such as encephalitis, syncope, migraine, cyclic vomiting syndrome and gastroenteritis; (d) the characteristic EEG findings suggested alternative diagnoses. Occipital spikes suggested "childhood epilepsy with occipital paroxysms" of Gastaut; multifocal spikes suggested symptomatic epilepsies with poor prognosis.
The veracity of Panayiotopoulos's initial descriptions has, over the last two decades, been confirmed in large and long-term studies from Europe, Japan and South America. The published database on which our knowledge of PS is now based includes over 800 cases of all races; there are few epilepsy syndromes which are better characterised."What emerges are a remarkably uniform clinical picture and a diagnosis which is strikingly useful in helping predict prognosis and dictate management."[48]
Autonomic status epilepticus is the more common type of nonfebrile status epilepticus in otherwise normal children and has been assessed in a consensus statement.[49]
References
- ↑ "Benign childhood occipital epilepsy, Panayiotopoulos type (Concept Id: C0393676) - MedGen - NCBI". www.ncbi.nlm.nih.gov. Archived from the original on 29 August 2023. Retrieved 27 August 2023.
- ↑ Panayiotopoulos CP. Panayiotopoulos syndrome: a common and benign childhood epileptic syndrome. London: John Libbey & Company; 2002.
- ↑ Ferrie C, Caraballo R, Covanis A, Demirbilek V, Dervent A, Kivity S et al. Panayiotopoulos syndrome: a consensus view. Dev Med Child Neurol 2006; 48(3):236-240.
- ↑ Panayiotopoulos CP. Panayiotopoulos syndrome: a common and benign childhood epileptic syndrome. London: John Libbey & Company; 2002.
- ↑ Koutroumanidis M. Panayiotopoulos Syndrome: An Important Electroclinical Example of Benign Childhood System Epilepsy. Epilepsia 2007; 48(6):1044-1053
- ↑ Caraballo R, Cersosimo R, Fejerman N. Panayiotopoulos syndrome: a prospective study of 192 patients. Epilepsia 2007; 48(6):1054-1061.
- ↑ Panayiotopoulos CP, Michael M, Sanders S, Valeta T, Koutroumanidis M. Benign childhood focal epilepsies: assessment of established and newly recognized syndromes. Brain 2008; 131(Pt 9):2264-2286.
- ↑ Koutroumanidis M, Ferrie CD, Valeta T, Sanders S, Michael M, Panayiotopoulos CP. Syncope-like epileptic seizures in Panayiotopoulos syndrome. Neurology 2012 July 31;79(5):463-7.
- ↑ Ferrie CD, Caraballo R, Covanis A, Demirbilek V, Dervent A, Fejerman N et al. Autonomic status epilepticus in Panayiotopoulos syndrome and other childhood and adult epilepsies: a consensus view. Epilepsia 2007; 48(6):1165-1172.
- ↑ Kanemura H, Sano F, Ohyama T, Aoyagi K, Sugita K, Aihara M. Sequential prefrontal lobe volume changes and cognitive dysfunctions in children with Panayiotopoulos syndrome presenting with status epilepticus. Epilepsy Res 2015; 112: 122-129..
- ↑ Verrotti A, Sebastiani M, Giordano L et al. Panayiotopoulos syndrome with convulsive status epilepticus at the onset: a long-term study. Seizure 2014; 23: 728-731.
- ↑ Cordelli DM, Aldrovandi A, Gentile V, Garone C, Conti S, Aceti A et al. Fever as a seizure precipitant factor in Panayiotopoulos syndrome: a clinical and genetic study. Seizure 2012 March;21(2):141-3.
- ↑ Cordelli DM, Aldrovandi A, Gentile V, Garone C, Conti S, Aceti A et al. Fever as a seizure precipitant factor in Panayiotopoulos syndrome: a clinical and genetic study. Seizure 2012 March;21(2):141-3.
- ↑ Zontek, Aneta; Paprocka, Justyna (31 May 2022). "Gastrointestinal and Autonomic Symptoms-How to Improve the Diagnostic Process in Panayiotopoulos Syndrome?". Children (Basel, Switzerland). 9 (6): 814. doi:10.3390/children9060814. ISSN 2227-9067. Retrieved 27 August 2023.
- ↑ Benarroch, EDUARDO E. (1 October 1993). "The Central Autonomic Network: Functional Organization, Dysfunction, and Perspective". Mayo Clinic Proceedings. 68 (10): 988–1001. doi:10.1016/S0025-6196(12)62272-1. ISSN 0025-6196. Retrieved 27 August 2023.
- ↑ Gonzalez-Duarte A, et al, Cardiovascular and neuroendocrine features of Panayiotopoulos syndrome in three siblings, Epilepsy Behav (2011), doi:10.1016/j.yebeh.2011.03.006
- ↑ Panayiotopoulos CP. Panayiotopoulos syndrome: a common and benign childhood epileptic syndrome. London: John Libbey & Company; 2002.
- ↑ Caraballo R, Cersosimo R, Fejerman N. Panayiotopoulos syndrome: a prospective study of 192 patients. Epilepsia 2007; 48(6):1054-1061.
- ↑ Panayiotopoulos CP, Michael M, Sanders S, Valeta T, Koutroumanidis M. Benign childhood focal epilepsies: assessment of established and newly recognized syndromes. Brain 2008; 131(Pt 9):2264-2286.
- ↑ Ohtsu M, Oguni H, Imai K, Funatsuka M, Osawa M. Early-onset form of benign childhood epilepsy with centro-temporal EEG foci - a different nosological perspective from panayiotopoulos syndrome. Neuropediatrics 2008; 39(1):14-19.
- ↑ Specchio N, Trivisano M, Claps D, Battaglia D, Fusco L, Vigevano F. Documentation of autonomic seizures and autonomic status epilepticus with ictal EEG in Panayiotopoulos syndrome. Epilepsy Behav 2010; 19(3):383-393.
- ↑ Saitoh N, Kanazawa O, Toyama J, Akasaka N, Kamimura T. Magnetoencephalographic findings of Panayiotopoulos syndrome with frontal epileptic discharges. Pediatr Neurol 2007;36:190-4.
- ↑ Saitoh N, Kanazawa O, Tohyama J, et al. Brain maturation-related spike localization in Panayiotopoulos syndrome: magnetoencephalographic study. Pediatr Neurol 2008;38(2):104-10.
- ↑ Koutroumanidis M, Ferrie CD, Valeta T, Sanders S, Michael M, Panayiotopoulos CP. Syncope-like epileptic seizures in Panayiotopoulos syndrome. Neurology 2012 July 31;79(5):463-7.
- ↑ Specchio N, Trivisano M, Claps D, Battaglia D, Fusco L, Vigevano F. Documentation of autonomic seizures and autonomic status epilepticus with ictal EEG in Panayiotopoulos syndrome. Epilepsy Behav 2010; 19(3):383-393.
- ↑ Panayiotopoulos CP. Panayiotopoulos syndrome: a common and benign childhood epileptic syndrome. London: John Libbey & Company; 2002.
- ↑ Berg AT, Berkovic SF, Brodie MJ, Buchhalter J, Cross HJ, Van Emde Boas W et al. Revised terminology and concepts for organization of seizures and epilepsies: Report of the ILAE Commission on Classification and Terminology, 2005-2009. Epilepsia 2010; 51:676-685.
- ↑ Ferrie CD, Caraballo R, Covanis A, Demirbilek V, Dervent A, Fejerman N et al. Autonomic status epilepticus in Panayiotopoulos syndrome and other childhood and adult epilepsies: a consensus view. Epilepsia 2007; 48(6):1165-1172.
- ↑ Ferrie CD, Caraballo R, Covanis A, Demirbilek V, Dervent A, Fejerman N et al. Autonomic status epilepticus in Panayiotopoulos syndrome and other childhood and adult epilepsies: a consensus view. Epilepsia 2007; 48(6):1165-1172.
- ↑ Covanis A. Panayiotopoulos Syndrome: A Benign Childhood Autonomic Epilepsy Frequently Imitating Encephalitis, Syncope, Migraine, Sleep Disorder, or Gastroenteritis. Pediatrics 2006; 118(4):e1237-e1243 doi:10.1542/peds.2006-0623.
- ↑ Ferrie C, Caraballo R, Covanis A, Demirbilek V, Dervent A, Kivity S et al. Panayiotopoulos syndrome: a consensus view. Dev Med Child Neurol 2006; 48(3):236-240.
- ↑ Panayiotopoulos CP, Michael M, Sanders S, Valeta T, Koutroumanidis M. Benign childhood focal epilepsies: assessment of established and newly recognized syndromes. Brain 2008; 131(Pt 9):2264-2286.
- ↑ Lacroix L, Fluss J, Gervaix A, Korff CM. Benzodiazepines in the acute management of seizures with autonomic manifestations: anticipate complications! Epilepsia 2011 October;52(10):e156-e159.
- ↑ Valeta T. Parental attitude, reaction and education in benign childhood focal seizures. In: Panayiotopoulos CP, editor. The Epilepsies: Seizures, Syndromes and Management. Oxford: Bladon Medical Publishing; 2005. 258-261.
- ↑ Panayiotopoulos CP. Panayiotopoulos syndrome: a common and benign childhood epileptic syndrome. London: John Libbey & Company; 2002.
- ↑ Koutroumanidis M. Panayiotopoulos Syndrome: An Important Electroclinical Example of Benign Childhood System Epilepsy. Epilepsia 2007; 48(6):1044-1053
- ↑ Caraballo R, Cersosimo R, Fejerman N. Panayiotopoulos syndrome: a prospective study of 192 patients. Epilepsia 2007; 48(6):1054-1061.
- ↑ Panayiotopoulos CP, Michael M, Sanders S, Valeta T, Koutroumanidis M. Benign childhood focal epilepsies: assessment of established and newly recognized syndromes. Brain 2008; 131(Pt 9):2264-2286.
- ↑ Specchio N, Trivisano M, Balestri M, Cappelletti S, Di Ciommo V, Gentile S et al. Panayiotopoulos syndrome: A Clinical, EEG and Neuropsychological Study of 93 Consecutive Patients. Epilepsia 2010; 51(10):2098-2107.
- ↑ Specchio N, Trivisano M, Balestri M, Cappelletti S, Di Ciommo V, Gentile S et al. Panayiotopoulos syndrome: A Clinical, EEG and Neuropsychological Study of 93 Consecutive Patients. Epilepsia 2010; 51(10):2098-2107.
- ↑ Caraballo R, Cersosimo R, Fejerman N. Panayiotopoulos syndrome: a prospective study of 192 patients. Epilepsia 2007; 48(6):1054-1061.
- ↑ Ferrie CD, Caraballo R, Covanis A, Demirbilek V, Dervent A, Fejerman N et al. Autonomic status epilepticus in Panayiotopoulos syndrome and other childhood and adult epilepsies: a consensus view. Epilepsia 2007; 48(6):1165-1172.
- ↑ Panayiotopoulos CP. Panayiotopoulos syndrome: a common and benign childhood epileptic syndrome. London: John Libbey & Company; 2002.
- ↑ Panayiotopoulos CP, Michael M, Sanders S, Valeta T, Koutroumanidis M. Benign childhood focal epilepsies: assessment of established and newly recognized syndromes. Brain 2008; 131(Pt 9):2264-2286.
- ↑ Panayiotopoulos CP. The Birth and Evolution of the Concept of Panayiotopoulos Syndrome. Epilepsia 2007; 48(6):1041-1043
- ↑ Panayiotopoulos CP. Vomiting as an ictal manifestation of epileptic seizures and syndromes. J Neurol Neurosurg Psychiatr 1988; 51(11):1448-1451.
- ↑ Ferrie CD, Livingston JH. Panayiotopoulos syndrome: learning lessons from atypical cases. Epileptic Disord 2010; 12(1):92-94
- ↑ Ferrie CD, Livingston JH. Panayiotopoulos syndrome: learning lessons from atypical cases. Epileptic Disord 2010; 12(1):92-94
- ↑ Ferrie CD, Caraballo R, Covanis A, Demirbilek V, Dervent A, Fejerman N et al. Autonomic status epilepticus in Panayiotopoulos syndrome and other childhood and adult epilepsies: a consensus view. Epilepsia 2007; 48(6):1165-1172.
External links
| Classification |
|---|