SCN1A



Sodium channel protein type 1 subunit alpha (SCN1A), is a protein which in humans is encoded by the SCN1A gene.[5][6][7][8]
Gene location
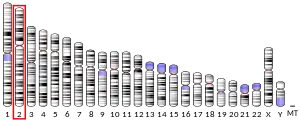
The SCN1A gene is located on chromosome 2 of humans, and is made up of 26 exons spanning a total length of 6030 nucleotide base pairs.[9][10] Alternative splicing of exon 5 gives rise to two alternate exons.[11] The promoter has been identified 2.5 kilobase pairs upstream of the transcription start site, and the 5'- untranslated exons may enhance expression of the SCN1A gene in SH-SY5Y cells, a human cell line derived from a neuroblastoma.[12]
Function
The vertebrate sodium channel is a voltage-gated ion channel essential for the generation and propagation of action potentials, chiefly in nerve and muscle. Voltage-sensitive sodium channels are heteromeric complexes consisting of a large central pore-forming glycosylated alpha subunit and 2 smaller auxiliary beta subunits. Functional studies have indicated that the transmembrane alpha subunit of the brain sodium channels is sufficient for expression of functional sodium channels.[13] Brain sodium channel alpha subunits form a gene subfamily with several structurally distinct isoforms clustering on chromosome 2q24, types I, II (Nav1.2), and III (Nav1.3). There are also several distinct sodium channel alpha subunit isoforms in skeletal and cardiac muscle (Nav1.4[14] and Nav1.5,[15] respectively).
The SCN1A gene codes for the alpha subunit of the voltage-gated sodium ion channel making it a member of ten paralogous gene families which code for the voltage-gated sodium transmembrane proteins NaV1.1. Within the family of genes which code for other portions of voltage-gated sodium channels, the SCN1A mutations were the first identified, since mutations to this gene caused epilepsy and febrile seizures.[16] Indeed, the SCN1A gene is one of the most commonly mutated genes in the human genome associated with epilepsy, which has given it the title of a 'super culprit gene'.[17] There are 900 distinct mutations reported for the SCN1A gene, approximately half of the reported mutations are truncations which result in no protein.[18][19] The remaining half of mutations are missense mutations, which are predicted to either cause loss-of-function or gain-of-function, though very few have been tested for functionality in the lab.[9]
Subtle differences in voltage-gated sodium ion channels can have devastating physiological effects and underlie abnormal neurological functioning.[20][21] Mutations to the SCN1A gene most often result in different forms of seizure disorders, the most common forms of seizure disorders are Dravet Syndrome (DS), Intractable childhood epilepsy with generalized tonic-clonic seizures (ICEGTC), and severe myoclonic epilepsy borderline (SMEB).[18] Clinically, 70-80% of patients with DS have identified mutations specific to the SCN1A gene, which are caused by de novo heterozygous mutations of the SCN1A gene.[22] There are currently two databases on SCN1A mutations, Infobase and the SCN1A variant database Archived 22 July 2019 at the Wayback Machine.
Mice with knock-in SCN1A mutations, who are model organisms for DS quickly develop seizures, indicative of a significant reduction in the function of NaV1.1.[10] It has been hypothesized that reduced sodium currents due to NaV1.1 mutations may cause hypoexcitability in GABAergic inhibitory interneurons leading to epilepsy.[12] Mice in both the homozygous and heterozygous states develop the seizure phenotype and ataxia. Though homozygous mice die on average during the second to third week of life and approximately 50% of heterozygous null mice survive into adulthood.[10][12][23]
Clinical significance
Mutations in the SCN1A gene cause inherited febrile seizures and GEFS+, type 2.[24][25][26][27]
Patent controversy
On 29 November 2008, The Sydney Morning Herald reported the first evidence of private intellectual property rights over human DNA[28] having adversely affected medical care. The Melbourne company Genetic Technologies (GTG) controls rights to the gene, and requires royalties for tests on the gene, which can help identify Dravet syndrome. Doctors on the Children's Hospital in Westmead, Australia have told journalists that they would test 50% more infants for the gene, if they could conduct the test on site.
Interactions
Nav1.1 has been shown to interact with syntrophin, alpha 1.[29]
See also
References
- 1 2 3 GRCh38: Ensembl release 89: ENSG00000144285 - Ensembl, May 2017
- 1 2 3 GRCm38: Ensembl release 89: ENSMUSG00000064329 - Ensembl, May 2017
- ↑ "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- ↑ "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- ↑ "Entrez Gene: SCN1A sodium channel, voltage-gated, type I, alpha subunit".
- ↑ Malo MS, Blanchard BJ, Andresen JM, Srivastava K, Chen XN, Li X, et al. (1994). "Localization of a putative human brain sodium channel gene (SCN1A) to chromosome band 2q24". Cytogenetics and Cell Genetics. 67 (3): 178–86. doi:10.1159/000133818. PMID 8062593.
- ↑ Ito M, Nagafuji H, Okazawa H, Yamakawa K, Sugawara T, Mazaki-Miyazaki E, et al. (January 2002). "Autosomal dominant epilepsy with febrile seizures plus with missense mutations of the (Na+)-channel alpha 1 subunit gene, SCN1A". Epilepsy Research. 48 (1–2): 15–23. doi:10.1016/S0920-1211(01)00313-8. PMID 11823106. S2CID 25555020.
- ↑ Catterall WA, Goldin AL, Waxman SG (December 2005). "International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels". Pharmacological Reviews. 57 (4): 397–409. doi:10.1124/pr.57.4.4. PMID 16382098. S2CID 7332624.
- 1 2 Meisler MH, O'Brien JE, Sharkey LM (June 2010). "Sodium channel gene family: epilepsy mutations, gene interactions and modifier effects". The Journal of Physiology. 588 (Pt 11): 1841–8. doi:10.1113/jphysiol.2010.188482. PMC 2901972. PMID 20351042.
- 1 2 3 Ogiwara I, Miyamoto H, Morita N, Atapour N, Mazaki E, Inoue I, et al. (May 2007). "Nav1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation". The Journal of Neuroscience. 27 (22): 5903–14. doi:10.1523/JNEUROSCI.5270-06.2007. PMC 6672241. PMID 17537961.
- ↑ Tate SK, Depondt C, Sisodiya SM, Cavalleri GL, Schorge S, Soranzo N, et al. (April 2005). "Genetic predictors of the maximum doses patients receive during clinical use of the anti-epileptic drugs carbamazepine and phenytoin". Proceedings of the National Academy of Sciences of the United States of America. 102 (15): 5507–12. Bibcode:2005PNAS..102.5507T. doi:10.1073/pnas.0407346102. PMC 556232. PMID 15805193.
- 1 2 3 Long YS, Zhao QH, Su T, Cai YL, Zeng Y, Shi YW, et al. (November 2008). "Identification of the promoter region and the 5'-untranslated exons of the human voltage-gated sodium channel Nav1.1 gene (SCN1A) and enhancement of gene expression by the 5'-untranslated exons". Journal of Neuroscience Research. 86 (15): 3375–81. doi:10.1002/jnr.21790. PMID 18655196. S2CID 33673297.
- ↑ Goldin AL, Snutch T, Lübbert H, Dowsett A, Marshall J, Auld V, et al. (October 1986). "Messenger RNA coding for only the alpha subunit of the rat brain Na channel is sufficient for expression of functional channels in Xenopus oocytes". Proceedings of the National Academy of Sciences of the United States of America. 83 (19): 7503–7. Bibcode:1986PNAS...83.7503G. doi:10.1073/pnas.83.19.7503. PMC 386747. PMID 2429308.
- ↑ George AL, Komisarof J, Kallen RG, Barchi RL (February 1992). "Primary structure of the adult human skeletal muscle voltage-dependent sodium channel". Annals of Neurology. 31 (2): 131–7. doi:10.1002/ana.410310203. PMID 1315496. S2CID 37892568.
- ↑ Gellens ME, George AL, Chen LQ, Chahine M, Horn R, Barchi RL, Kallen RG (January 1992). "Primary structure and functional expression of the human cardiac tetrodotoxin-insensitive voltage-dependent sodium channel". Proceedings of the National Academy of Sciences of the United States of America. 89 (2): 554–8. Bibcode:1992PNAS...89..554G. doi:10.1073/pnas.89.2.554. PMC 48277. PMID 1309946.
- ↑ Escayg, A., MacDonald, B. T., Meisler, M. H., Baulac, S., Huberfeld, G., An-Gourfinkel, I., … Malafosse, A. (2000). Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nature Genetics, 24(4), 343–345. doi:10.1038/74159
- ↑ Lossin, C. (2009). A catalog of SCN1A variants. Brain and Development, 31(2), 114–130. doi:10.1016/j.braindev.2008.07.011
- 1 2 Fujiwara, T., Sugawara, T., Mazaki‐Miyazaki, E., Takahashi, Y., Fukushima, K., Watanabe, M., … Inoue, Y. (2003). Mutations of sodium channel α subunit type 1 (SCN1A) in intractable childhood epilepsies with frequent generalized tonic–clonic seizures. Brain, 126(3), 531–546. doi:10.1093/brain/awg053
- ↑ Ohmori, I., Kahlig, K. M., Rhodes, T. H., Wang, D. W., & George, A. L. (2006). Nonfunctional SCN1A Is Common in Severe Myoclonic Epilepsy of Infancy. Epilepsia, 47(10), 1636–1642. doi:10.1111/j.1528-1167.2006.00643.x
- ↑ Kohrman, D. C., Smith, M. R., Goldin, A. L., Harris, J., & Meisler, M. H. (1996). A Missense Mutation in the Sodium Channel Scn8a Is Responsible for Cerebellar Ataxia in the Mouse Mutant jolting. Journal of Neuroscience, 16(19), 5993–5999.
- ↑ Bulman, D. E. (1997). Phenotype Variation and Newcomers in Ion Channel Disorders. Human Molecular Genetics, 6(10), 1679–1685. doi:10.1093/hmg/6.10.1679
- ↑ Claes, L., Del-Favero, J., Ceulemans, B., Lagae, L., Van Broeckhoven, C., & De Jonghe, P. (2001). De Novo Mutations in the Sodium-Channel Gene SCN1A Cause Severe Myoclonic Epilepsy of Infancy. American Journal of Human Genetics, 68(6), 1327–1332.
- ↑ Yu, F. H., Mantegazza, M., Westenbroek, R. E., Robbins, C. A., Kalume, F., Burton, K. A., … Catterall, W. A. (2006). Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nature Neuroscience, 9(9), 1142–1149. doi:10.1038/nn1754
- ↑ Escayg A, MacDonald BT, Meisler MH, Baulac S, Huberfeld G, An-Gourfinkel I, et al. (April 2000). "Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2". Nature Genetics. 24 (4): 343–5. doi:10.1038/74159. PMID 10742094. S2CID 29543172.
- ↑ Spampanato J, Escayg A, Meisler MH, Goldin AL (October 2001). "Functional effects of two voltage-gated sodium channel mutations that cause generalized epilepsy with febrile seizures plus type 2". The Journal of Neuroscience. 21 (19): 7481–90. doi:10.1523/jneurosci.21-19-07481.2001. PMC 6762922. PMID 11567038.
- ↑ Nabbout R, Gennaro E, Dalla Bernardina B, Dulac O, Madia F, Bertini E, et al. (June 2003). "Spectrum of SCN1A mutations in severe myoclonic epilepsy of infancy". Neurology. 60 (12): 1961–7. doi:10.1212/01.wnl.0000069463.41870.2f. PMID 12821740. S2CID 604240.
- ↑ Lossin C. "SCN1A infobase". Retrieved 30 October 2009.
compilation of genetic variations in the SCN1A gene that alter the expression or function of Nav1.1
- ↑ Robotham J (29 November 2008). "Sick babies denied treatment in DNA row –". National News. Sidney Morning Herald – smh.com.au. Retrieved 3 December 2008.
- ↑ Gee SH, Madhavan R, Levinson SR, Caldwell JH, Sealock R, Froehner SC (January 1998). "Interaction of muscle and brain sodium channels with multiple members of the syntrophin family of dystrophin-associated proteins". The Journal of Neuroscience. 18 (1): 128–37. doi:10.1523/jneurosci.18-01-00128.1998. PMC 6793384. PMID 9412493.
Further reading
- Lerche H, Jurkat-Rott K, Lehmann-Horn F (2001). "Ion channels and epilepsy". American Journal of Medical Genetics. 106 (2): 146–59. doi:10.1002/ajmg.1582. PMID 11579435.
- Isom LL (January 2002). "The role of sodium channels in cell adhesion". Frontiers in Bioscience. 7 (1–3): 12–23. doi:10.2741/isom. PMID 11779698.
- Kanai K, Hirose S, Oguni H, Fukuma G, Shirasaka Y, Miyajima T, et al. (July 2004). "Effect of localization of missense mutations in SCN1A on epilepsy phenotype severity". Neurology. 63 (2): 329–34. doi:10.1212/01.wnl.0000129829.31179.5b. PMID 15277629. S2CID 36070893.
- Oguni H, Hayashi K, Osawa M, Awaya Y, Fukuyama Y, Fukuma G, et al. (2004). "Severe myoclonic epilepsy in infancy: clinical analysis and relation to SCN1A mutations in a Japanese cohort". Advances in Neurology. 95: 103–17. PMID 15508916.
- Mulley JC, Scheffer IE, Petrou S, Dibbens LM, Berkovic SF, Harkin LA (June 2005). "SCN1A mutations and epilepsy". Human Mutation. 25 (6): 535–42. doi:10.1002/humu.20178. PMID 15880351. S2CID 19148287.
- Catterall WA, Goldin AL, Waxman SG (December 2005). "International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels". Pharmacological Reviews. 57 (4): 397–409. doi:10.1124/pr.57.4.4. PMID 16382098. S2CID 7332624.
- Lu CM, Han J, Rado TA, Brown GB (May 1992). "Differential expression of two sodium channel subtypes in human brain". FEBS Letters. 303 (1): 53–8. doi:10.1016/0014-5793(92)80476-W. PMID 1317301. S2CID 29330026.
- Goldin AL, Snutch T, Lübbert H, Dowsett A, Marshall J, Auld V, et al. (October 1986). "Messenger RNA coding for only the alpha subunit of the rat brain Na channel is sufficient for expression of functional channels in Xenopus oocytes" (PDF). Proceedings of the National Academy of Sciences of the United States of America. 83 (19): 7503–7. Bibcode:1986PNAS...83.7503G. doi:10.1073/pnas.83.19.7503. PMC 386747. PMID 2429308.
- Malo MS, Blanchard BJ, Andresen JM, Srivastava K, Chen XN, Li X, et al. (1994). "Localization of a putative human brain sodium channel gene (SCN1A) to chromosome band 2q24". Cytogenetics and Cell Genetics. 67 (3): 178–86. doi:10.1159/000133818. PMID 8062593.
- Peiffer A, Thompson J, Charlier C, Otterud B, Varvil T, Pappas C, et al. (October 1999). "A locus for febrile seizures (FEB3) maps to chromosome 2q23-24". Annals of Neurology. 46 (4): 671–8. doi:10.1002/1531-8249(199910)46:4<671::AID-ANA20>3.0.CO;2-5. PMID 10514109. S2CID 33638988.
- Wallace RH, Scheffer IE, Barnett S, Richards M, Dibbens L, Desai RR, et al. (April 2001). "Neuronal sodium-channel alpha1-subunit mutations in generalized epilepsy with febrile seizures plus". American Journal of Human Genetics. 68 (4): 859–65. doi:10.1086/319516. PMC 1275639. PMID 11254444.
- Escayg A, Heils A, MacDonald BT, Haug K, Sander T, Meisler MH (April 2001). "A novel SCN1A mutation associated with generalized epilepsy with febrile seizures plus--and prevalence of variants in patients with epilepsy". American Journal of Human Genetics. 68 (4): 866–73. doi:10.1086/319524. PMC 1275640. PMID 11254445.
- Whitaker WR, Faull RL, Waldvogel HJ, Plumpton CJ, Emson PC, Clare JJ (March 2001). "Comparative distribution of voltage-gated sodium channel proteins in human brain". Brain Research. Molecular Brain Research. 88 (1–2): 37–53. doi:10.1016/S0169-328X(00)00289-8. PMID 11295230.
- Claes L, Del-Favero J, Ceulemans B, Lagae L, Van Broeckhoven C, De Jonghe P (June 2001). "De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy". American Journal of Human Genetics. 68 (6): 1327–32. doi:10.1086/320609. PMC 1226119. PMID 11359211.
- Sugawara T, Mazaki-Miyazaki E, Ito M, Nagafuji H, Fukuma G, Mitsudome A, et al. (August 2001). "Nav1.1 mutations cause febrile seizures associated with afebrile partial seizures". Neurology. 57 (4): 703–5. doi:10.1212/wnl.57.4.703. PMID 11524484. S2CID 45138036.
- Abou-Khalil B, Ge Q, Desai R, Ryther R, Bazyk A, Bailey R, et al. (December 2001). "Partial and generalized epilepsy with febrile seizures plus and a novel SCN1A mutation". Neurology. 57 (12): 2265–72. doi:10.1212/wnl.57.12.2265. PMID 11756608. S2CID 26448714.
- Ito M, Nagafuji H, Okazawa H, Yamakawa K, Sugawara T, Mazaki-Miyazaki E, et al. (January 2002). "Autosomal dominant epilepsy with febrile seizures plus with missense mutations of the (Na+)-channel alpha 1 subunit gene, SCN1A". Epilepsy Research. 48 (1–2): 15–23. doi:10.1016/S0920-1211(01)00313-8. PMID 11823106. S2CID 25555020.
External links
- GeneReviews/NCBI/NIH/UW entry on Familial Hemiplegic Migraine
- GeneReviews/NCBI/NIH/UW entry on SCN1A-Related Seizure Disorders
- SCN1A+protein,+human at the U.S. National Library of Medicine Medical Subject Headings (MeSH)
PDB gallery | |
|---|---|
|

This article incorporates text from the United States National Library of Medicine, which is in the public domain.