Mammary secretory carcinoma
| Mammary secretory carcinoma | |
|---|---|
| Other names | Secretory carcinoma of the breast |
| Specialty | Breast surgery, Surgical oncology |
| Complications | Very rare cases may become aggressive cancers |
| Usual onset | Adult females, less commonly adult males or female and male children |
| Duration | Very slow growing |
| Causes | Formation of an ETV6-NTRK3 fusion gene |
| Prognosis | good to excellent |
| Frequency | rare |
| Deaths | rare |
Mammary secretory carcinoma (MSC), also termed secretory carcinoma of the breast, is a rare form of the breast cancers.[1] MSC usually affects women but in a significant percentage of cases also occurs in men and children.[2] Indeed, McDvitt and Stewart first described MSC in 1966 and termed it juvenile breast carcinoma because an increased number of cases were at that time diagnosed in juvenile females.[3] MSC is the most common form of breast cancer in children,[4] representing 80% of childhood breast cancers,[5] although it accounts for less than 0.15% of all breast cancers.[6] (In the pediatric population, about 65% of all breast malignancies are carcinomas.[5])
In 1980, Tavaosoli and Norris renamed juvenile breast carcinoma as "secretory carcinoma of the breast" based on its characteristic microscopic appearance[7] of having eosinophilic (i.e. red or pink) extracellular secretions when its tissues were stained with the H&E stain.[8] MSC has sometimes been termed secretory carcinoma without reference to its breast cancer location. However, the term secretory carcinoma has also been used to name tumors with the microscopic appearance of MSC that are localized in the salivary glands (now termed mammary analogue secretory carcinoma),[9] thyroid gland (now termed salivary gland–type carcinoma of the thyroid),[10] or skin (now termed secretory carcinoma of the skin or primary cutaneous mammary analog secretory carcinoma).[11][12] Mammary secretory carcinoma is used here to distinguish it from these formerly termed secretory carcinomas.
MSCs typically develop as ductal breast tumors that have invaded the tissue around their ducts of origin, may have spread to nearby sentinel lymph nodes or axillary lymph nodes but have rarely metastasized (i.e. spread) to distant tissues.[6] They are usually small, slow-growing, painless,[5] well-circumscribed, movable breast masses.[4] MSC tumors typically have distinctive microscopic features and tumor cells that carry a characteristic genetic abnormality that appears to underlie their development and/or progression.[2]
Surgical removal has been considered the mainstay of treatment for localized MSCs tumors. The rare cases of MSC tumors that have metastasized to distal tissues have been treated with chemotherapy and radiation therapy but have shown little to no responses to these treatments. Emerging studies suggest that drug therapy targeted at the genetic abnormality in MSC tumor cells may be a more effective treatment for the latter tumors.[13]
Presentation
In a review study of 190 individuals diagnosed with MSC, patient ages ranged from 2 to 96 years (median age: 56 years); 5% of cases were aged less than 21 years; the male to female ratio was 1 to 30; the tumors were in the left, right, and both breasts in 103, 85 and 2 cases, respectively; and the tumors were usually located on a breast's left, outer quadrant.[1] (In younger patients, these tumors more often occur beneath the areola, i.e. pigmented region around the breast's nipple.[14]) In a subgroup of 99 patients on which there was sufficient information, average tumor sizes were 2.3 centimeters (cm.) with 29.3% of these patients evidencing lymph node metastases.[1] This study did not have patients who presented with metastasis to tissues beyond their sentinel lymph nodes. In a review study of 44 individuals with MSC, 41 patients were female and 3 were male; patient ages ranged between 4 and 76 years (median age: 48 years) with 4 patients being adolescents; patient tumor sizes ranged from 1.5 to 10 cm. (mean size: 3.5 cm.) (Individual cases of MSC have had tumor sizes as large as 16 cm.[4]) and 15 patients presented with lymph node metastases. While none of these patients presented with metastasis beyond sentinel lymph nodes, follow up studies over an average of 93.4 months identified 4 patients with metastases to the bone, 1 patient with metastases to the liver, and 1 patient with unspecified distant metastases. Five of these patients died of their disease.[15]
In a review study 12 female and 2 male patients 8 to 81 years (mean age: 48 years), 8 patients presented with a palpable mass, 4 presented with a mass detected on screening mammography, and 2 patients lacked information on this; 8 tumors were located in the left and 6 in the right breast; and 2 of the 10 patents tested by biopsy had sentinel lymph node metastases. One of these patients presented with distant tissue metastases, 12 re-presented with distant metastasis within 5–240 months of their initial diagnoses and treatments, and one lacked information on this. Eight of these patients died from their disease 7 to 240 months (mean survival time: 74.6 months) after their initial diagnosis, 4 were alive with their disease 7 to 84 months after their initial diagnosis, and 2 had no information on this. The metastases, which often were to multiple sites, were located in the lung (9 cases), liver (4 cases), bone (4 cases), skin (2 cases), kidney (1 case), mediastinum (1 case), pancreas (1 case), and/or lung's pleura (1 case).[8] Rare cases of MSC have presented in individuals who have or had and been treated for papillomatosis of breasts; this benign breast diseases is often associated with other breast carcinomas, the by far most common of which is MSC.[5]
As of 2019, only 6 cases presented with purely in situ (i.e. totally localized tumors that have not invaded tissues outside of the breasts' ducts) tumors (as reported in the English literature). The tumor tissues in all of these cases had at least one area with a papillary microscopic histopathology (see Pathology section).[16]
Pathology
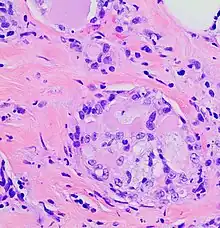
The microscopic histopathology of H&E-stained MSC tumors generally shows well-circumscribed solid tumors with multiple microcysts[13] and sheets of epithelial cells containing numerous vacuoles in their cytoplasm, signet ring cells (i.e. cells with one large cytoplasmic vacuole termed a globule), and extracellular droplet-like secretions resembling the cytoplasmic globules.[2] The sheets of cells are separated by bands of connective tissue.[5] In occasional cases, the cells in MSC tumors are arranged in a papillary (i.e. finger-like)[17] or tubular[14] pattern. The extracellular microcysts and intracellular vacuoles and globules stain red when stained with the H&E stain, consist of sulfated glycosaminoglycans and mucin-containing material,[4] and are identified using periodic acid-Shiff, PAD diastase, or Alician blue stains.[15] The term “secretory” for MSC is in recognition that the tumor cells vacuoles and globules are secreted extracellularly to give the tissues their microcystic appearance.[8] At least 2 cases of MSC had sarcoma-like malignant cells in portions of their tumors and a rapidly metastasizing disease.[18][19]
Immunohistochemistry
Immohistochemical analyses of MSC tumors detects cells that: in less than 50% of patients (including a male child[4]) express estrogen and/or progesterone receptors; in rare cases express HER2/neu receptors;[14] and in most cases express S100 protein, mammaglobin protein,[17] and cytokeratin 5 or 6 (as detected using cytokeratin 5/6 antibodies).[16] In a study of 44 patients, MSC tumor cells expressed the estrogen receptor, progesterone receptor, HER2/Neu receptor, p53 tumor protein, and S100 protein in 21, 23, 16, 12, and 42 patients, respectively.[15]
Genetic abnormality
MSC tumors carry a mutation, i.e. a balanced genetic translocation, in which part of the NTRK3 gene located in band two, five sub-band 2 (see chromosme banding) on the q (i.e. long) arm of chromosome 15 is fused to a part of the ETV6 gene located in band one, three sub-band 2 on chromosome 12.[14] The fusion gene product, ETV6-NTRK3, encodes a chimeric mutant protein, termed ETV6-NTRK3.[6] This fusion protein and its encoding ETV6-NTRK3 fusion gene are detected in the tumor cells of most (e.g. 88.6%[13] to 100%[8]) cases of MSC. The NTRK3 portion of ETV6-NTRK3 protein has up-regulated tyrosine kinase activity. This activity stimulates two signaling pathways, the PI3K/AKT/mTOR and MAPK/ERK pathways, that promote cell proliferation, cell survival and, it is thought, thereby contribute to the development and/or progression of MSC. These pathways may also be responsible for stimulating the development of the vacuoles and globules seen in MSC tumors.[6] The ETV6-NTRK3 fusion gene and its ETV6-NTRK3 chimeric protein product are also detected in the tumor cells of mammary analogue secretory carcinoma of salivary glands,[20][21] salivary gland–type carcinomas of the thyroid,[22] and secretory carcinoma of the skin.[12] This translocation is the sole genetic mutation reported in most MSC tumors; is a hallmark characteristic genetic abnormality in MSC tumor cells;[1] is used to confirm the diagnosis of MSC;[17] and has led to treating malignant MSC tumors with a drug, Larotrectinib, that inhibits the tyrosine kinase activity of the NTRK3 portion of the ETV6-NTRK3 fusion protein.[6]
Diagnosis
The diagnosis of MSC tumors rests on their occurrence in breast ductal tissue, their distinct microscopic histopathological features[2] plus, particularly in tumors showing a papillary histopathology,[17] the presence of in the tumor of cells that express the ETV6-NTRK3 fusion gene and/or its ETV6-NTRK3 chimeric protein.[1][15][16][17] While the ETV6-NTRK3 gene fusion occurs in many other tumor types (e.g. inflammatory myofibroblastic tumors, the cellular variant of congenital mesoblastic nephroma, and radiation-induced papillary thyroid carcinoma), these tumors' clinical presentations and histopathology findings distinguish them from MSC tumors.[13]
Treatment
There have been no controlled studies that have evaluated the many different strategies that have been used to treat MSC tumors.[16] Studies have suggested that: 1) MSC tumor sizes less than 2[13] or 3[15] centimeters in widest dimension, tumors with circumscribed margins, tumors with no evidence of axillary lymph node metastasis,[15] tumors in individuals who are less than 20[13] or 30[14] years old at diagnosis, and/or tumors that are purely in situ[16] have more favorable prognoses; 2) MSC tumors with high rates of proliferation, tumors greater than 2 to 3 centimeters in widest dimension,[13][15] tumors that contain areas of sarcoma-like cells,[19] tumors in individuals older than 20 or 30 years,[13][14] and/or tumors that have spread to distant tissues[8] have less favorable prognoses.
In general, surgery has been considered the best treatment for MSC tumors but there is no consensus on the extent of this surgery.[15] Conservative surgery or simple mastectomy with biopsy of sentinel lymph nodes has been recommended for patients with MSC tumors that have more favorable prognoses. Removal of adjacent mammary tissue to the tumor to insure that all invasive tissue is removed has also been recommended since leaving tumor tissue behind after surgery runs a higher risk of local recurrences.[1] Tumors with unfavorable prognostic features have been treated with radical mastectomy. Since MSC tumors grow slowly, carry relatively good prognoses, and if metastasizing to distant tissues usually do so only 10 to 20 years after,[14] there is no strong evidence for the benefit of adjuvant chemotherapy and/or radiotherapy[4] A retrospective study of 190 patients with MSC that had not metastasized to distant tissue found that breast-conserving surgery plus radiotherapy gave significantly better survival times than simple mastectomy.[5] In general, conservative surgery, modified radical mastectomy, and radical mastectomy have been the most frequent procedures in adults while simple mastectomy, local excision with sentinel lymph node biopsy, and complete axillary dissection have been recommended as adequate treatment for children with MSC.[15] Further studies are needed to confirm that radiotherapy improves one or more of the latter treatment regimens.[5] In all cases, long-term follow-up examination, e.g. for at least 20 years after diagnoses, is strongly recommended.[1][14]
The rare cases of MSC tumors that spread to distant tissues on their initial presentation or after prior treatment have been treated with various chemotherapy regimens. Responses to these regiments have been poor to nil.[8][13][14] Recently, three individuals (two juvenile females and one 26 year old male) with ETV6-NTRK3 fusion gene-harboring MSC tumors that metastasized to distant tissues were treated with Larotrectinib, a drug which attacks the genetic abnormality underlying this disease. The drug is an orally available selective NTRKi inhibitor that blocks the NTRK3 protein's tyrosine kinase activity. It has produced good, fast-developing responses in the three reported MSC cases.[6][13][16] However, these three responses were partial, i.e. some tumor tissue remained after treatment.[23] Further studies are needed on a much larger number of individuals with MSC to define the efficacy of Larotrectinib, the duration of its effects, and the efficacy of other MTRK3 protein inhibitors such as Entrectinib.[6][23]
Prognosis
Overall, MSC has a favorable prognosis.[5][23] A study of 190 individuals treated for MSC that had not spread to distant tissues reported survival rates (excluding individuals dying of other causes) at 5, 10, and 20 years after initial treatment of 95.79, 93.16, and 93.16%, respectively.[1] The uncommon cases of MSC which have metastasize to distant tissues may also have prolonged survival times[8] but some have proven to be fatal with shorter survival times (e.g. 1 year after diagnosis).[13][15][19] Larotrectinib therapy may improve the survival times in individuals with MSC tumors that have poorer prognoses.[23]
References
- 1 2 3 4 5 6 7 8 Gong P, Xia C, Yang Y, Lei W, Yang W, Yu J, Ji Y, Ren L, Ye F (July 2021). "Clinicopathologic profiling and oncologic outcomes of secretory carcinoma of the breast". Scientific Reports. 11 (1): 14738. Bibcode:2021NatSR..1114738G. doi:10.1038/s41598-021-94351-w. PMC 8289843. PMID 34282256.
- 1 2 3 4 Carretero-Barrio I, Santón A, Caniego Casas T, López Miranda E, Reguero-Callejas ME, Pérez-Mies B, Benito A, Palacios J (February 2022). "Cytological and molecular characterization of secretory breast carcinoma". Diagnostic Cytopathology. 50 (7): E174–E180. doi:10.1002/dc.24945. PMC 9303577. PMID 35156343. S2CID 246813006.
- ↑ McDivitt RW, Stewart FW (January 1966). "Breast carcinoma in children". JAMA. 195 (5): 388–90. doi:10.1001/jama.1966.03100050096033. PMID 4285563.
- 1 2 3 4 5 6 Novochadlo Klüppel E, Rodrigues da Costa L, Marquetto Tognolo C, do Nascimento A, Grignet Ribeiro M, Girardi Fachin C (2020). "Secretory breast carcinoma in a male child: Case report and literature review". International Journal of Surgery Case Reports. 73: 310–314. doi:10.1016/j.ijscr.2020.07.040. PMC 7394736. PMID 32736235.
- 1 2 3 4 5 6 7 8 Knaus ME, Grabowksi JE (August 2021). "Pediatric Breast Masses: An Overview of the Subtypes, Workup, Imaging, and Management". Advances in Pediatrics. 68: 195–209. doi:10.1016/j.yapd.2021.05.006. PMID 34243852. S2CID 235786044.
- 1 2 3 4 5 6 7 Loo SK, Yates ME, Yang S, Oesterreich S, Lee AV, Wang XS (May 2022). "Fusion-associated carcinomas of the breast: Diagnostic, prognostic, and therapeutic significance". Genes, Chromosomes & Cancer. 61 (5): 261–273. doi:10.1002/gcc.23029. PMC 8930468. PMID 35106856.
- ↑ Tavassoli FA, Norris HJ (May 1980). "Secretory carcinoma of the breast". Cancer. 45 (9): 2404–13. doi:10.1002/1097-0142(19800501)45:9<2404::aid-cncr2820450928>3.0.co;2-8. PMID 6445777. S2CID 9074530.
- 1 2 3 4 5 6 7 Hoda RS, Brogi E, Pareja F, Nanjangud G, Murray MP, Weigelt B, Reis-Filho JS, Wen HY (August 2019). "Secretory carcinoma of the breast: clinicopathologic profile of 14 cases emphasising distant metastatic potential". Histopathology. 75 (2): 213–224. doi:10.1111/his.13879. PMC 6646069. PMID 31012486.
- ↑ Vasanthi V, Ramadoss R (2021). "Secretory carcinoma of salivary gland - A systematic review of pediatric case reports and case series". Journal of Oral and Maxillofacial Pathology. 25 (2): 327–331. doi:10.4103/0973-029X.325236. PMC 8491356. PMID 34703129.
- ↑ Baloch ZW, Asa SL, Barletta JA, Ghossein RA, Juhlin CC, Jung CK, LiVolsi VA, Papotti MG, Sobrinho-Simões M, Tallini G, Mete O (March 2022). "Overview of the 2022 WHO Classification of Thyroid Neoplasms". Endocrine Pathology. 33 (1): 27–63. doi:10.1007/s12022-022-09707-3. PMID 35288841. S2CID 247440666.
- ↑ Amin SM, Beattie A, Ling X, Jennings LJ, Guitart J (November 2016). "Primary Cutaneous Mammary Analog Secretory Carcinoma With ETV6-NTRK3 Translocation". The American Journal of Dermatopathology. 38 (11): 842–845. doi:10.1097/DAD.0000000000000590. PMID 27763904. S2CID 46372654.
- 1 2 Taniguchi K, Yanai H, Kaji T, Kubo T, Ennishi D, Hirasawa A, Yoshino T (August 2021). "Secretory carcinoma of the skin with lymph node metastases and recurrence in both lungs: A case report". Journal of Cutaneous Pathology. 48 (8): 1069–1074. doi:10.1111/cup.14028. PMID 33882152. S2CID 233352244.
- 1 2 3 4 5 6 7 8 9 10 11 Tang H, Zhong L, Jiang H, Zhang Y, Liang G, Chen G, Xie G (June 2021). "Secretory carcinoma of the breast with multiple distant metastases in the brain and unfavorable prognosis: a case report and literature review". Diagnostic Pathology. 16 (1): 56. doi:10.1186/s13000-021-01115-1. PMC 8223364. PMID 34162406.
- 1 2 3 4 5 6 7 8 9 Banerjee N, Banerjee D, Choudhary N (2021). "Secretory carcinoma of the breast, commonly exhibits the features of low grade, triple negative breast carcinoma- A Case report with updated review of literature". Autopsy & Case Reports. 11: e2020227. doi:10.4322/acr.2020.227. PMC 8101654. PMID 34277491.
- 1 2 3 4 5 6 7 8 9 10 Li L, Wu N, Li F, Li L, Wei L, Liu J (February 2019). "Clinicopathologic and molecular characteristics of 44 patients with pure secretory breast carcinoma". Cancer Biology & Medicine. 16 (1): 139–146. doi:10.20892/j.issn.2095-3941.2018.0035. PMC 6528460. PMID 31119054.
- 1 2 3 4 5 6 Yang Y, Wang Z, Pan G, Li S, Wu Y, Liu L (August 2019). "Pure secretory carcinoma in situ: a case report and literature review". Diagnostic Pathology. 14 (1): 95. doi:10.1186/s13000-019-0872-7. PMC 6706916. PMID 31443715.
- 1 2 3 4 5 Kulka J, Madaras L, Floris G, Lax SF (January 2022). "Papillary lesions of the breast". Virchows Archiv. 480 (1): 65–84. doi:10.1007/s00428-021-03182-7. PMC 8983543. PMID 34734332.
- ↑ Srinivas V, Harjai MM, Subramanya H, Rajaram T, Rai R (October 2004). "Carcinosarcoma of the Breast With An Unusual Secretory Carcinoma as the Carcinomatous Component". Medical Journal, Armed Forces India. 60 (4): 410–2. doi:10.1016/S0377-1237(04)80027-3. PMC 4923432. PMID 27407689.
- 1 2 3 Xu J, Weisman P (June 2021). "Dedifferentiated secretory breast carcinoma with fibrosarcomatous features harboring an ETV6-NTRK3 fusion in both components". Genes, Chromosomes & Cancer. 60 (6): 447–451. doi:10.1002/gcc.22929. PMID 33342011. S2CID 229341868.
- ↑ Janik S, Faisal M, Marijić B, Grasl S, Grasl MC, Heiduschka G, Erovic BM (March 2022). "Prognostic factors in mammary analogue secretory carcinomas of the parotid gland: Systematic review and meta-analysis". Head & Neck. 44 (3): 792–804. doi:10.1002/hed.26971. PMID 34964195. S2CID 245539420.
- ↑ Aref-Eshghi E, Lin F, Li MM, Zhong Y (November 2021). "The oncogenic roles of NTRK fusions and methods of molecular diagnosis". Cancer Genetics. 258–259: 110–119. doi:10.1016/j.cancergen.2021.10.005. PMID 34710798. S2CID 239217797.
- ↑ Chu YH, Dias-Santagata D, Farahani AA, Boyraz B, Faquin WC, Nosé V, Sadow PM (November 2020). "Clinicopathologic and molecular characterization of NTRK-rearranged thyroid carcinoma (NRTC)". Modern Pathology. 33 (11): 2186–2197. doi:10.1038/s41379-020-0574-4. PMC 7584778. PMID 32457407.
- 1 2 3 4 Mortensen L, Ordulu Z, Dagogo-Jack I, Bossuyt V, Winters L, Taghian A, Smith BL, Ellisen LW, Kiedrowski LA, Lennerz JK, Bardia A, Spring LM (October 2021). "Locally Recurrent Secretory Carcinoma of the Breast with NTRK3 Gene Fusion". The Oncologist. 26 (10): 818–824. doi:10.1002/onco.13880. PMC 8488779. PMID 34176200.