Necroptosis
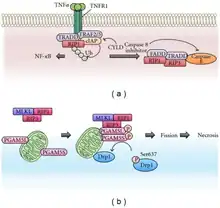
Necroptosis is a programmed form of necrosis, or inflammatory cell death.[1] Conventionally, necrosis is associated with unprogrammed cell death resulting from cellular damage or infiltration by pathogens, in contrast to orderly, programmed cell death via apoptosis. The discovery of necroptosis showed that cells can execute necrosis in a programmed fashion and that apoptosis is not always the preferred form of cell death. Furthermore, the immunogenic nature of necroptosis favors its participation in certain circumstances, such as aiding in defence against pathogens by the immune system. Necroptosis is well defined as a viral defense mechanism, allowing the cell to undergo "cellular suicide" in a caspase-independent fashion in the presence of viral caspase inhibitors to restrict virus replication.[2] In addition to being a response to disease, necroptosis has also been characterized as a component of inflammatory diseases such as Crohn's disease, pancreatitis, and myocardial infarction.[3][4]
The signaling pathway responsible for carrying out necroptosis is generally understood. TNFα leads to stimulation of its receptor TNFR1. TNFR1 binding protein TNFR-associated death protein TRADD and TNF receptor-associated factor 2 TRAF2 signals to RIPK1 which recruits RIPK3 forming the necrosome also named ripoptosome.[2] Phosphorylation of MLKL by the ripoptosome drives oligomerization of MLKL, allowing MLKL to insert into and permeabilize plasma membranes and organelles.[5][6] Integration of MLKL leads to the inflammatory phenotype and release of damage-associated molecular patterns (DAMPs), which elicit immune responses.
Function
Necroptosis is specific to vertebrates and may have originated as an additional defense to pathogens. Necroptosis also acts as an alternative "fail-safe" cell death pathway in cases where cells are unable to undergo apoptosis, such as during viral infection in which apoptosis signaling proteins are blocked by the virus.
In innate immunity
Cell suicide is an effective means of stemming the spread of a pathogen throughout an organism. In apoptotic responses to infection, the contents of an infected cell (including the pathogen) are contained and engulfed by phagocytosis. Some pathogens, such as human cytomegalovirus, express caspase inhibitors that arrest the apoptotic machinery of the host cell.[7] The caspase-independence of necroptosis allows the cell to bypass caspase activation, decreasing the time during which the pathogen can inhabit the cell.
Toll-like receptors (TLRs) can also signal to the necrosome, leading to necroptosis. TLRs are a class of receptors that function in the innate immune system to recognize conserved components of pathogens, such as flagellin.[2]
In contrast to apoptosis
In apoptosis, extrinsic signaling via cell surface receptors or intrinsic signaling by release of cytochrome c from mitochondria leads to caspase activation. Proteolytic degradation of the cell's interior culminates with the packaging of the cell's remains into apoptotic bodies, which are degraded and recycled by phagocytosis. Unlike in apoptosis, necrosis and necroptosis do not involve caspase activation. Necrotic cell death culminates in leakage of cell contents into the extracellular space, in contrast to the organized disposal of cellular contents into apoptotic bodies.[8]
Process
As in all forms of necrotic cell death, cells undergoing necroptosis rupture and leak their contents into the intercellular space. Unlike in necrosis, permeabilization of the cell membrane during necroptosis is tightly regulated. While many of these mechanisms and components of the pathway are still being uncovered, the major steps of necroptotic signaling have been outlined in recent years. First, extrinsic stimulus through the TNF receptor by TNFα signals the recruitment of the TNF receptor-associated death domain (TRADD) which in turn recruits RIPK1. In the absence of active Caspase 8, RIPK1 and RIPK3 auto- and transphosphorylate each other, leading to the formation of a microfilament-like complex called the necrosome.[2] The necrosome then activates the pro-necroptotic protein MLKL via phosphorylation. MLKL actuates the necrosis phenotype by inserting into the bilipid membranes of organelles and plasma membrane leading to expulsion of cellular contents into the extracellular space.[5][6] The inflammatory rupturing of the cell releases Damage Associated Molecular Patterns (DAMPs) into the extracellular space. Many of these DAMPs remain unidentified, however, the "find me" and "eat me" DAMP signals are known to recruit immune cells to the damaged/infected tissue.[8] Necrotic cells are cleared from the immune system by a mechanism called pinocytosis, or cellular drinking, which is mediated by macropinosomes, a subcellular component of macrophages. This process is in contrast to removal of apoptotic cells by the immune system in which cells are removed via phagocytosis, or cellular eating.
Co-regulation of necroptosis and apoptosis
Recent studies have shown substantial interplay between the apoptosis and necroptosis pathways. At multiple stages of their respective signalling cascades, the two pathways can regulate each other. The best characterized example of this co-regulation is the ability of caspase 8 to inhibit the formation of the necrosome by cleaving RIPK1. Conversely, caspase 8 inhibition of necroptosis can be bypassed by the necroptotic machinery through the anti-apoptotic protein cFLIP which inactivates caspase 8 through formation of a heterodimer.[4]
Many components of the two pathways are also shared. The Tumor Necrosis Factor Receptor can signal for both apoptosis and necroptosis. The RIPK1 protein can also signal for both apoptosis and necroptosis depending on post-translational modifications mediated by other signalling proteins. Furthermore, RIPK1 can be regulated by cellular inhibitor of apoptosis proteins 1 and 2 (cIAP1, cIAP2) which polyubiquitinate RIPK1 leading to cell survival through downstream NF-kB signalling. cIAP1 and cIAP2 can also be regulated by the pro-apoptotic protein SMAC (second mitochondria-derived activator of caspases) which can cleave cIAP1 and cIAP2 driving the cell towards an apoptotic death.[2]
Targeting of organelles
Cells can undergo necroptosis in response to perturbed homeostasis in specific circumstances. In response to DNA damage, the RIPK1 and RIPK3 are phosphorylated and lead to deterioration of the cell in the absence of caspase activation. The necrosome inhibits the adenine nucleotide translocase in mitochondria to decrease cellular ATP levels.[8] Uncoupling of the mitochondrial electron transport chain leads to additional mitochondrial damage and opening of the mitochondrial permeability transition pore, which releases mitochondrial proteins into the cytosol. The necrosome also causes leakage of lysosomal digestive enzymes into the cytoplasm by induction of reactive oxygen species by JNK, sphingosine production, and calpain activation by calcium release.
Medical relevance
Necroptosis has been implicated in the pathology of many types of acute tissue damage, including myocardial infarction, stroke, ischemia-reperfusion injury. In addition, necroptosis is noted to contribute to atherosclerosis, pancreatitis, inflammatory bowel disease, neurodegeneration, and some cancers.[9]
In solid-organ transplantation, ischemia-reperfusion injury can occur when blood returns to tissue for the first time in the transplant recipient. A major contributor to tissue damage results from activation of regulated necroptosis, which could include contributions from both necroptosis and mitochondrial permeability transition. Treatment with the drug cyclosporine, which represses the mitochondrial permeability transition effector Cyclophilin D, improves tissue survival primarily by inhibiting necrotic cell death, rather than its additional function as an immunosuppressant.[4]
Necroptosis based-therapy
Recently, necroptosis-based cancer therapy, using a distinctive molecular pathway for regulation of necroptosis, has been suggested as an alternative method to overcome apoptosis-resistance. For instance, necroptotic cells release highly immunogenic DAMPs, initiating adaptive immunity. These dying cells can also activate NF-κB to express cytokines, recruiting macrophages.[10] As of 2018 little is known about negative regulators of necroptosis, but CHIP, cFLIP and FADD appear to be potential targets for necroptosis based therapy.[10]
References
- ↑ Nirmala JG, Lopus M (April 2020). "Cell death mechanisms in eukaryotes". Cell Biology and Toxicology. 36 (2): 145–164. doi:10.1007/s10565-019-09496-2. PMID 31820165.
- 1 2 3 4 5 Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P (February 2014). "Regulated necrosis: the expanding network of non-apoptotic cell death pathways". Nature Reviews. Molecular Cell Biology. 15 (2): 135–47. doi:10.1038/nrm3737. PMID 24452471. S2CID 13919892.
- ↑ Günther C, Martini E, Wittkopf N, Amann K, Weigmann B, Neumann H, Waldner MJ, Hedrick SM, Tenzer S, Neurath MF, Becker C (September 2011). "Caspase-8 regulates TNF-α-induced epithelial necroptosis and terminal ileitis". Nature. 477 (7364): 335–9. Bibcode:2011Natur.477..335G. doi:10.1038/nature10400. PMC 3373730. PMID 21921917.
- 1 2 3 Linkermann A, Green DR (January 2014). "Necroptosis". The New England Journal of Medicine. 370 (5): 455–65. doi:10.1056/nejmra1310050. PMC 4035222. PMID 24476434.
- 1 2 Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, Wang FS, Wang X (April 2014). "Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3". Molecular Cell. 54 (1): 133–146. doi:10.1016/j.molcel.2014.03.003. PMID 24703947.
- 1 2 Su L, Quade B, Wang H, Sun L, Wang X, Rizo J (October 2014). "A plug release mechanism for membrane permeation by MLKL". Structure. 22 (10): 1489–500. doi:10.1016/j.str.2014.07.014. PMC 4192069. PMID 25220470.
- ↑ Mocarski ES, Upton JW, Kaiser WJ (December 2011). "Viral infection and the evolution of caspase 8-regulated apoptotic and necrotic death pathways". Nature Reviews. Immunology. 12 (2): 79–88. doi:10.1038/nri3131. PMC 4515451. PMID 22193709.
- 1 2 3 Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G (October 2010). "Molecular mechanisms of necroptosis: an ordered cellular explosion". Nature Reviews. Molecular Cell Biology. 11 (10): 700–14. doi:10.1038/nrm2970. PMID 20823910. S2CID 7424865.
- ↑ Zhou W, Yuan J (November 2014). "Necroptosis in health and diseases". Seminars in Cell & Developmental Biology. 35: 14–23. doi:10.1016/j.semcdb.2014.07.013. PMID 25087983.
- 1 2 Razaghi A, Heimann K, Schaeffer PM, Gibson SB (January 2018). "Negative regulators of cell death pathways in cancer: perspective on biomarkers and targeted therapies". Apoptosis. 23 (2): 93–112. doi:10.1007/s10495-018-1440-4. PMID 29322476. S2CID 3424489.