Peters-plus syndrome
| Peters-plus syndrome | |
|---|---|
| Other names: Krause–van Schooneveld–Kivlin syndrome | |
 | |
| This condition is inherited in an autosomal recessive manner | |
Peters-plus syndrome or Krause–Kivlin syndrome is a hereditary syndrome defined by Peters' anomaly, dwarfism and intellectual disability.[1][2]
Signs and symptoms
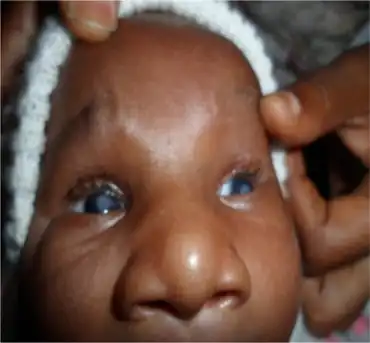
Features of this syndrome include Peters' anomaly, corneal opacity, central defect of Descemet's membrane, and shallow anterior chamber with synechiae between the iris and cornea.
Craniofacial abnormalities commonly seen in patients with PPS include hypertelorism, ear malformations, micrognathia, round face and broad neck, and cleft lip and palette.[1]
Infants are commonly born small for gestational age and have delayed growth. It is associated with short limb dwarfism and mild to severe intellectual disability and autism spectrum disorder.[1]
Cause
The pattern of inheritance of Peters-plus is autosomal recessive, where both parents are heterozygous they can produce a child with the syndrome. The B3GALTL[3] (now called B3GLCT) gene codes for the enzyme beta 3-glucosyltransferase (B3Glc-T).[4]
The beta 3-glucosyltransferase enzyme is responsible for glycosylation, the attachment of sugars to proteins, which through this modification allows for performance of a wider variety of functions. The mutations of the B3GLCT gene in affected individuals results in loss-of-function of the beta 3-glucosyltransferase enzyme. The result of this disruption in glycosylation is a change to the secondary structure of the mRNA. These mutations of the B3GLCT gene lead to the production of an abnormally short, nonfunctional version of the beta 3-glucosyltransferase (B3Glc-T) enzyme, which disrupts glycosylation.[5]
The phenotypic effects of the B3GLCT mutations result in a triad of well known phenotypes; Peters anomaly (also classified as anterior segment defects, a defect in the anterior cornea), short stature, brachydactyly, in addition to several other less frequently observed phenotypes.[6] A study of 55 patients with Peters-plus-related phenotypes, but lacking the most common combination (Peters anomaly, short stature, and brachydactyly), revealed none of those cases displayed mutation in the B3GLCT gene. Thus PPS-like signs and symptoms, when they occur independently of each other, provide strong evidence that the B3GLCT gene mutation is in fact responsible for actual cases Peters-plus syndrome.[7]
Diagnosis
The evaluation of this condition, Peters-plus syndrome, is based on the following:[8]
- Clinical exam
- Genetic testing
- Ophthalmologic exam
- Imaging studies
Management
The treatment for this condition is based on the symptom(s) presented.[8]
History
Krause–van Schooneveld–Kivlin syndrome is listed as a "rare disease" by the Office of Rare Diseases (ORD) of the National Institutes of Health (NIH).[9]
It was characterized in 1984 by van Schooneveld.[10]
See also
References
- 1 2 3 Smith's Recognizable Patterns of Human Malformation (7 ed.). Elsevier. 2013. pp. 752–795.
- ↑ "Krause–Kivlin syndrome'". U. S. National Library of Medicine Congenital Syndromes Database Archives. NIH. Archived from the original on 15 January 2015. Retrieved 3 August 2012.
- ↑ Weh, Eric; Reis, Linda M.; Tyler, Rebecca C.; Bick, David; Rhead, William J.; Wallace, Stephanie; McGregor, Tracy L.; Dills, Shelley K.; Chao, Mei-Chyn; Murray, Jeffrey C.; Semina, Elena V. (August 2014). "Novel B3GALTL mutations in classic Peters Plus syndrome and lack of mutations in a large cohort of patients with similar phenotypes". Clinical Genetics. 86 (2): 142–148. doi:10.1111/cge.12241. ISSN 0009-9163. PMC 4103962. PMID 23889335.
- ↑ Heinonen, Taisto Y. K.; Maki, Markku (2009). "Peters'-plus syndrome is a congenital disorder of glycosylation caused by a defect in the beta1,3-glucosyltransferase that modifies thrombospondin type 1 repeats". Annals of Medicine. 41 (1): 2–10. doi:10.1080/07853890802301975. ISSN 1365-2060. PMID 18720094. S2CID 33364600.
- ↑ Siala, Olfa; Belguith, Neila; Kammoun, Hassen; Kammoun, Bourane; Hmida, Nedia; Chabchoub, Imen; Hchicha, Mongia; Fakhfakh, Faiza (2012-10-01). "Two Tunisian patients with Peters plus syndrome harbouring a novel splice site mutation in the B3GALTL gene that modulates the mRNA secondary structure". Gene. 507 (1): 68–73. doi:10.1016/j.gene.2012.06.052. ISSN 0378-1119. PMID 22759511. Archived from the original on 2021-11-19. Retrieved 2021-09-13.
- ↑ Weh, Eric; Takeuchi, Hideyuki; Muheisen, Sanaa; Haltiwanger, Robert S.; Semina, Elena V. (2017-09-19). "Functional characterization of zebrafish orthologs of the human Beta 3-Glucosyltransferase B3GLCT gene mutated in Peters Plus Syndrome". PLOS ONE. 12 (9): e0184903. Bibcode:2017PLoSO..1284903W. doi:10.1371/journal.pone.0184903. ISSN 1932-6203. PMC 5604996. PMID 28926587.
- ↑ Weh, E.; Reis, L. M.; Tyler, R. C.; Bick, D.; Rhead, W. J.; Wallace, S.; McGregor, T. L.; Dills, S. K.; Chao, M.-C.; Murray, J. C.; Semina, E. V. (August 2014). "Novel B3GALTL mutations in classic Peters plus syndrome and lack of mutations in a large cohort of patients with similar phenotypes". Clinical Genetics. 86 (2): 142–148. doi:10.1111/cge.12241. ISSN 1399-0004. PMC 4103962. PMID 23889335.
- 1 2 "Peters plus syndrome | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Archived from the original on 14 April 2021. Retrieved 19 November 2021.
- ↑ "Peters plus syndrome". Genetic and Rare Diseases Information Center (GARD). NIH. Archived from the original on 28 November 2012. Retrieved 3 August 2012.
- ↑ van Schooneveld MJ, Delleman JW, Beemer FA, Bleeker-Wagemakers EM (December 1984). "Peters'-plus: a new syndrome". Ophthalmic Paediatr Genet. 4 (3): 141–5. doi:10.3109/13816818409006113. PMID 6443615.
External links
| Classification | |
|---|---|
| External resources |
|
- GeneReviews/NCBI/NIH/UW entry on Peters Plus Syndrome Archived 2010-05-28 at the Wayback Machine
- OMIM entries on Peters Plus syndrome Archived 2021-11-19 at the Wayback Machine
- Peters Anomaly at eMedicine on eMedicine