Tricho-hepato-enteric syndrome
| Tricho-hepato-enteric syndrome | |
|---|---|
| Other names: Intractable diarrhea of infancy with facial dysmorphism[1] | |
.jpg.webp) | |
| Typical facial abnormalities with prominent forehead and cheeks, broad nasal root and wide-spaced eyes. Abnormal hairs are woolly, easily removed and poorly pigmented. | |
Tricho-hepato-enteric syndrome (THE), also known as syndromic or phenotypic diarrhea, is an extremely rare congenital bowel disorder which manifests itself as intractable diarrhea in infants with intrauterine growth retardation, hair and facial abnormalities.[2] Many also have liver disease and abnormalities of the immune system.[2] The associated malabsorption leads to malnutrition and failure to thrive.[2]
It is thought to be a genetic disorder with an autosomal recessive inheritance pattern, although responsible genes have not been found and the exact cause remains unknown. Prognosis is poor; many patients die before the age of 5 (mainly from infections or cirrhosis), although most patients nowadays survive with intravenous feeding (parenteral nutrition).
Symptoms and signs
Tricho-hepato-enteric syndrome is one particular form of intractable diarrhea of infancy, presenting typically in the first month of life. These babies were usually born small for their age and continue to experience failure to thrive, usually with a final short stature. Typical facial features include prominent forehead and cheeks, a broad nasal root and widely spaced eyes (hypertelorism). Their hairs are woolly, easily removed and poorly pigmented. Liver disease is mainly present as cirrhosis or fibrosis, and staining might reveal high iron content of the liver cells (consistent with hemochromatosis).[3] Most evaluated patients had some degree of decrease in intelligence.
Genetics
The syndrome appears to be due to mutations in the gene tetratricopeptide repeat domain 37 (TTC37) which encodes the protein Thespin or the SKIV2L gene.[4][5] This gene is expressed is in the adrenal gland, amniotic fluid, bladder, blood, bone, bone marrow, brain, cervix, connective tissue, ear, epididymis, eye, heart, intestine, kidney, liver, lung, lymph nodes, mammary glands, mouth, muscle, nerve, oesophagus, ovary, pancreas, pharynx, placenta, prostate, pituitary gland, salivary gland, testis, thyroid, tonsil, thymus, trachea, skin, uterus, spleen, spinal cord, stomach and vascular tissue. It is also expressed in ascites and various embryonic tissues. It is expressed at high level in the intestine, lung, lymph nodes, pituitary and vascular tissues. This gene is also known as KIAA0372, MGC32587 and TPR repeat protein 37.
This gene is located on the Crick (minus) strand of the long arm of chromosome 5 (5q15). The gene is 91,113 bases in length and encodes a protein of 1564 amino acid residues with twenty tetratricopeptide repeats. It has 43 exons of which exons 1, 2 and 3 are non coding. The predicted molecular weight of the protein is 175.486 kiloDaltons and its predicted pI is 7.47. Its function is unknown but it may have adenylate cyclase activity and calcium- and calmodulin-responsive adenylate cyclase activity. A homolog has been identified in the frog (Xenopus tropicalis), the mouse (Mus musculus) and the rat (Rattus norvegicus). In the mouse this gene is located on chromosome 13.
Diagnosis
.jpg.webp)
Facial features
The typical facial features are low-set ears, prominent eyes with hypertelorism, broad flat nose, prominent forehead and large mouth.
Liver
There may be fibrosis with bile duct proliferation, occasional giant cells and regenerative parenchymal nodules. Siderosis is common.
Small bowel
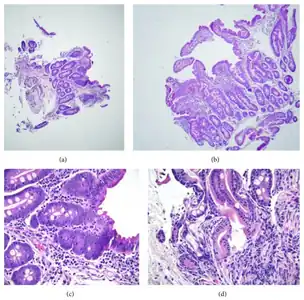 a,b)Moderate/severe shortening of most villi c)focal superficial epithelial d) crypt architectural changes.
a,b)Moderate/severe shortening of most villi c)focal superficial epithelial d) crypt architectural changes..jpg.webp) Small intestine biopsy of a patient with THE syndrome showing severe villous atrophy with intense mononuclear cell infiltration in the lamina propria. (Courtesy of Prof. Michel Peuchmaur, Hôpital Robert Debré, Paris, France)
Small intestine biopsy of a patient with THE syndrome showing severe villous atrophy with intense mononuclear cell infiltration in the lamina propria. (Courtesy of Prof. Michel Peuchmaur, Hôpital Robert Debré, Paris, France)
Microscopic examination of a biopsy of the small bowel in these patients shows villous atrophy with low or no mononuclear cell infiltration of the lamina propria nor specific abnormalities involving the epithelium. The amount of villous atrophy does not explain the severity of the diarrhea.
Studies of enterocyte brush-border ion transporter proteins (sodium-hydrogen exchanger 2, sodium-hydrogen exchanger 3, aquaporin 7, sodium iodide symporter and hydrogen potassium adenosine triphosphatase) showed reduced expression or mislocalization in all patients with different profiles for each.[6]
Hair
Microscopic analysis of the hair shows twisted hairs of unequal size and different shapes (pili torti, aniso- and poikilotrichosis), longitudinal breaks and breaks located at nodes (trichorrhexis nodosa). Scanning electron microscopy might reveal hair budding (trichorrhexis blastysis). Biochemical analysis may reveal sulfur-deficient brittle hair (trichothiodystrophy; note that disulfide bonds determine hair waviness).
Platelets
Platelets may be enlarged. The membrane surface connected canalicular system is disrupted with prominent tubules and small membranous vesicles. Alpha granules may be missing from the platelets. Despite these abnormalities there is no increased tendency to bleed in this syndrome.
Other
More than 90% of patients present immune defects. Low immunoglobulin level, a defect in antibody production after vaccination, monoclonal hyper IgA, and low lymphocyte count have been reported. In these cases, some patients may need immunoglobulin supplementation.[7]
Treatment
No specific treatment or cure exists. Affected children usually need total parenteral nutrition through a central venous catheter. Further worsening of liver damage should however be avoided if possible. Diarrhea will likely continue even though food stops passing through the gastrointestinal system.[8] They can subsequently be managed with tube feeding, and some may be weaned from nutritional support during adolescence.
Epidemiology
Tricho-hepato-enteric syndrome is estimated to affect 1 in 300,000 to 400,000 live births in Western Europe. This syndrome was first reported in 1982 with a report on 2 siblings,[9] and as of 2008 there were around 25 published cases in medical journals. There seem to be no racial differences in its occurrence. It might be more common, as many genetic diseases, in areas with high levels of consanguinity.
Footnotes
- ↑ Fabre A, André N, Breton A, Broué P, Badens C, Roquelaure B (March 2007). "Intractable diarrhea with "phenotypic anomalies" and tricho-hepato-enteric syndrome: two names for the same disorder". Am. J. Med. Genet. A. 143 (6): 584–8. doi:10.1002/ajmg.a.31634. PMID 17318842. S2CID 39567209.
- 1 2 3 Goulet O, Vinson C, Roquelaure B, Brousse N, Bodemer C, Cézard JP (2008). "Syndromic (phenotypic) diarrhea in early infancy". Orphanet J Rare Dis. 3: 6. doi:10.1186/1750-1172-3-6. PMC 2279108. PMID 18304370.
- ↑ Verloes A, Lombet J, Lambert Y, et al. (February 1997). "Tricho-hepato-enteric syndrome: further delineation of a distinct syndrome with neonatal hemochromatosis phenotype, intractable diarrhea, and hair anomalies". Am. J. Med. Genet. 68 (4): 391–5. doi:10.1002/(SICI)1096-8628(19970211)68:4<391::AID-AJMG3>3.0.CO;2-P. PMID 9021008.
- ↑ Fabre A, Martinez-Vinson C, Roquelaure B, Missirian C, André N, Breton A, Lachaux A, Odul E, Colomb V, Lemale J, Cézard JP, Goulet O, Sarles J, Levy N, Badens C (2011). "Novel mutations in TTC37 associated with tricho-hepato-enteric syndrome" (PDF). Hum Mutat. 32 (3): 277–281. doi:10.1002/humu.21420. PMID 21120949. S2CID 22834121. Archived (PDF) from the original on 2022-01-11. Retrieved 2021-07-13.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - ↑ Vardi I, Barel O, Sperber M, Schvimer M, Nunberg M, Field M, Ouahed J, Marek-Yagel D, Werner L, Haberman Y, Lahad A, Anikster Y, Rechavi G, Barshack I, McElwee JJ, Maranville J, Somech R, Snapper SB, Weiss B, Shouval DS (May 2018). "Genetic and Structural Analysis of a SKIV2L Mutation Causing Tricho-hepato-enteric Syndrome". Dig Dis Sci. 63 (5): 1192–1199. doi:10.1007/s10620-018-4983-x. PMC 6167312. PMID 29484573.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - ↑ Hartley JL, Zachos NC, Dawood B, Donowitz M, Forman J, Pollitt RJ, Morgan NV, Tee L, Gissen P, Kahr WH, Knisely AS, Watson S, Chitayat D, Booth IW, Protheroe S, Murphy S, de Vries E, Kelly DA, Maher ER (2010). "Mutations in TTC37 cause trichohepatoenteric syndrome (phenotypic diarrhea of infancy)". Gastroenterology. 138 (7): 2388–2398. doi:10.1053/j.gastro.2010.02.010. PMC 3166659. PMID 20176027.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - ↑ Fabre, A., Martinez-Vinson, C., Goulet, O. et al. Syndromic diarrhea/Tricho-hepato-enteric syndrome. Orphanet J Rare Dis 8, 5 (2013). https://doi.org/10.1186/1750-1172-8-5 Archived 2022-01-24 at the Wayback Machine
- ↑ Girault D, Goulet O, Le Deist F, et al. (July 1994). "Intractable infant diarrhea associated with phenotypic abnormalities and immunodeficiency". J. Pediatr. 125 (1): 36–42. doi:10.1016/S0022-3476(94)70118-0. PMID 8021782.
- ↑ Stankler L, Lloyd D, Pollitt RJ, Gray ES, Thom H, Russell G (March 1982). "Unexplained diarrhoea and failure to thrive in 2 siblings with unusual facies and abnormal scalp hair shafts: a new syndrome". Arch. Dis. Child. 57 (3): 212–6. doi:10.1136/adc.57.3.212. PMC 1627586. PMID 7073301.
References
- Goulet O, Vinson C, Roquelaure B, Brousse N, Bodemer C, Cézard JP (2008). "Syndromic (phenotypic) diarrhea in early infancy". Orphanet J Rare Dis. 3: 6. doi:10.1186/1750-1172-3-6. PMC 2279108. PMID 18304370.
External links
| Classification | |
|---|---|
| External resources |
|