Variably protease-sensitive prionopathy
| Variably protease-sensitive prionopathy | |
|---|---|
 | |
| Protease senstitive and resistant prionopathies coexisting in this immunohistological test | |
| Symptoms | Dementia, behavioral and psychiatric symptoms, ataxia, parkinsonism |
| Usual onset | 70 years |
| Causes | Prion |
| Differential diagnosis | Alzheimer's disease |
| Treatment | Supportive |
| Prognosis | Average survival 24 months |
| Frequency | 2-3 per 100 million people |
Variably protease-sensitive prionopathy (VPSPr) (formerly known as Protease Sensitive Prionopathy) is a sporadic prion protein disease first described in an abstract for a conference on prions in 2006, and this study was published in a 2008 report on 11 cases. The study was conducted by Gambetti P., Zou W.Q., and coworkers from the United States National Prion Disease Pathology Surveillance Center.[1][2] It was first identified as a distinct disease in 2010 by Zou W.Q. and coworkers from the United States National Prion Disease Pathology Surveillance Center.[3]
VPSPr is very rare, occurring in just 2 or 3 out of every 100 million people.[4] As of 2018, fourteen cases have been reported in the UK.[5] It has similarities to Creutzfeldt–Jakob disease, but clinical manifestations differ somewhat, and the abnormal prion protein (PrP) is less resistant to digestion by proteases; some variants are more sensitive to proteases than others, hence the name: variably protease-sensitive.
Patients present with behavioral and psychiatric symptoms, speech deficits (aphasia and/or dysarthria) and progressive cognitive and motor decline (dementia, ataxia, parkinsonism, psychosis, aphasia and mood disorder). The average age at onset is 70 years, and the duration of survival is 24 months. About 40% of patients have a family history of dementia. Like CJD, it can be mistaken for Alzheimer's dementia.
Diagnosis is difficult, as pathognomonic signs on MRI such as cortical ribboning or hockey stick sign, periodic sharp wave complexes on EEG, and tests for 14-3-3 protein and tau protein are usually not helpful, and no mutations have been observed in the coding region of the PrP gene, unlike CJD and Variant CJD.[6] The diagnosis can be made on pathological examination. There are unique microscopic and immunohistochemical features, and the prions cannot be digested using proteases. Because 8 out of 10 patients had a positive family history of dementia in the original study, a genetic cause was suspected. Some have suggested the disease may be a sporadic form of Gerstmann–Sträussler–Scheinker syndrome (GSS).[7]
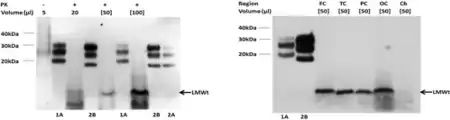
In 2013, Zou W.Q. and coworkers revealed that the peculiar protease-resistant PrP (PrPres) originally found in VPSPr is also detectable in the brain of patients with a genetic CJD linked to PrP Valine (V) to isoleucine (I) mutation at residue 180 (PrPV180I); moreover, they found that the pathological PrP from both VPSPr and gCJDPrPV180I shares a similar glycoform-selective prion formation mechanism.[8,9] The authors further demonstrated that the protease-resistant PrPres from both VPSPr and gCJDV180I lacks the PrP species glycosylated at the first N-linked glycosylation site at residue 181 and they proposed that the deficiency in PrP glycosylation may be involved in the pathogenesis of the two conditions. In 2014, Gambetti P., Zou W.Q., and coworkers found that approximately 54% of mice inoculated with VPSPr brain homogenates exhibited histopathologic lesions and 34% harbored abnormal PrP similar to that of VPSPr on the first passage but no prion disease was detected on the second passage,[10] suggesting that the infectivity of the pathological PrP from VPSPr is lower compared to that from the most common sporadic CJD.
See also
References
- ↑ Gambetti P, Dong Z, Yuan J, et al. (June 2008). "A novel human disease with abnormal prion proteining sensitive to protease". Ann. Neurol. 63 (6): 697–708. doi:10.1002/ana.21420. PMC 2767200. PMID 18571782.
- ↑ "The Merck Manual - Variably Protease-Sensitive Prionopathy (VPSPr)". Merck Sharp & Dohme Corp., a subsidiary of Merck & Co. Archived from the original on 16 November 2014. Retrieved 16 November 2014.
- ↑ Zou, W.; Puoti, G.; Xiao, X.; Yuan, J.; Qing, L.; Cali, I.; Shimoji, M.; Langeveld, J.; Castellani, R.; Notari, S.; Crain, B.; Schmidt, R. E.; Geschwind, M.; Dearmond, S. J.; Cairns, N. J.; Dickson, D.; Honig, L.; Torres, J. M.; Mastrianni, J.; Capellari, S.; Giaccone, G.; Belay, E. D.; Schonberger, L. B.; Cohen, M.; Perry, G.; Kong, Q.; Parchi, P.; Tagliavini, F.; Gambetti, P. (2010). "Variably protease-sensitive prionopathy: A new sporadic disease of the prion protein". Annals of Neurology. 68 (2): 162–172. doi:10.1002/ana.22094. PMC 3032610. PMID 20695009.
- ↑ "Variably Protease-Sensitive Prionopathy (VPSPr) - Neurologic Disorders - Merck Manuals Professional Edition". Merck Manuals Professional Edition. Archived from the original on 2018-02-03. Retrieved 2018-03-04.
- ↑ "Creutzfeldt - Jakob disease in the UK (By calendar year)" (PDF). Archived (PDF) from the original on 4 July 2018. Retrieved 4 July 2018.
- ↑ Will R, Head M (June 2008). "A new prionopathy". Ann. Neurol. 63 (6): 677–8. doi:10.1002/ana.21447. PMID 18570344. S2CID 33632289.
- ↑ Nonno, Romolo; Notari, Silvio; Di Bari, Michele Angelo; Cali, Ignazio; Pirisinu, Laura; d'Agostino, Claudia; Cracco, Laura; Kofskey, Diane; Vanni, Ilaria; Lavrich, Jody; Parchi, Piero; Agrimi, Umberto; Gambetti, Pierluigi (2019). "Variable Protease-Sensitive Prionopathy Transmission to Bank Voles". Emerging Infectious Diseases. 25 (1): 73–81. doi:10.3201/eid2501.180807. PMC 6302590. PMID 30561322.
- 8. Xiao X, Yuan J, Haïk S, Cali I, Zhan YA, Moudjou M, Li B, Laplanche JL, Laude H, Langeveld J, Gambetti P, Kitamoto T, Kong Q, Brandel JP, Cobb BA, Petersen RB & Zou WQ. Glycoform-selective prion formation in sporadic and familial forms of prion disease. PLoS ONE, 2013; 8:e58786.
- 9. Zou, WQ, Gambetti P, Xiao X, Yuan J, Langeveld J & Pirisinu L. Prions in variably protease-sensitive prionopathy: An update. Pathogens 2013; 2(3): 457-471.
- 10. Notari S, Xiao X, Espinosa JC, Cohen Y, Qing L, Aguilar-Calvo P, Kofskey D, Cali I, Cracco L, Kong Q, Torres JM, Zou W & Gambetti P. Transmission characteristics of variably protease-sensitive prionopathy. Emerg Infect Dis 2014, 20:2006-14.
External links
| Classification | |
|---|---|
| External resources |
|
- Caroline Parkinson (13 August 2010). "Brain disease could affect more people, research finds". BBC News. Archived from the original on 9 November 2020. Retrieved 15 July 2023.