Viral pathogenesis
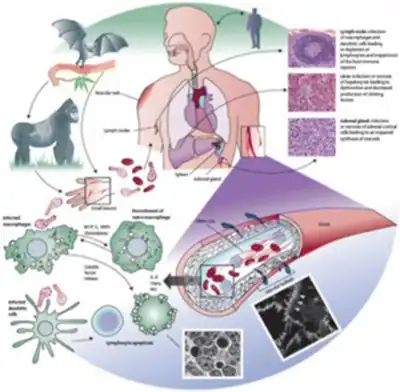
Viral pathogenesis is the process and mechanisms by which viruses cause diseases in their target hosts, often at the cellular or molecular level. It is a specialized field of study in virology.[1]
Pathogenesis is a qualitative description of the process by which an initial infection causes disease.[2] Viral disease is the sum of the effects of viral replication on the host and the host's subsequent immune response against the virus.[3] Viruses are able to initiate infection, disperse throughout the body, and replicate due to specific virulence factors.[2]
There are several factors that affect pathogenesis. Some of these factors include virulence characteristics of the virus that is infecting. In order to cause disease, the virus must also overcome several inhibitory effects present in the host. Some of the inhibitory effects include distance, physical barriers and host defenses. These inhibitory effects may differ among individuals due to the inhibitory effects being genetically controlled.
Viral pathogenesis is affected by various factors: (1) transmission, entry and spread within the host, (2) tropism, (3) virus virulence and disease mechanisms, (4) host factors and host defense.[4]
Mechanisms of infection
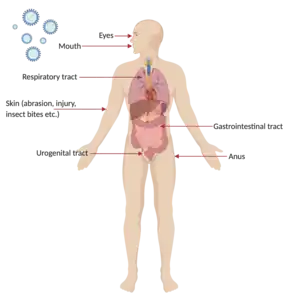
Viruses need to establish infections in host cells in order to multiply. For infections to occur, the virus has to hijack host factors and evade the host immune response for efficient replication. Viral replication frequently requires complex interactions between the virus and host factors that may result in deleterious effects in the host, which confers the virus its pathogenicity.[5]
Important steps of a virus life cycle that shape pathogenesis
- Transmission from a host with an infection to a second host
- Entry of the virus into the body
- Local replication in susceptible cells
- Dissemination and spread to secondary tissues and target organs
- Secondary replication in susceptible cells
- Shedding of the virus into the environment
- Onward transmission to third host
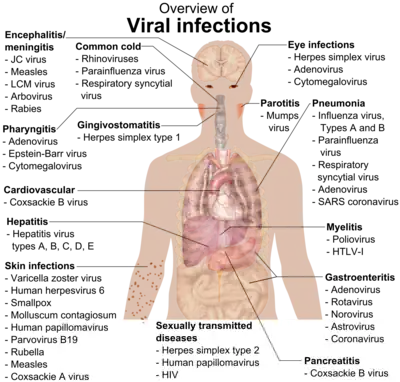
Primary transmission
Three requirements must be satisfied to ensure successful infection of a host. Firstly, there must be sufficient quantity of virus available to initiate infection. Cells at the site of infection must be accessible, in that their cell membranes display host-encoded receptors that the virus can exploit for entry into the cell, and the host anti-viral defense systems must be ineffective or absent.[3][5]
Entry to host
Viruses causing disease in humans often enter through the mouth, nose, genital tract, or through damaged areas of skin, so cells of the respiratory, gastrointestinal, skin and genital tissues are often the primary site of infection.[2][7][4] Some viruses are capable of transmission to a mammalian fetus through infected germ cells at the time of fertilization, later in pregnancy via the placenta, and by infection at birth.[2]
Local replication and spread
Following initial entry to the host, the virus hijacks the host cell machinery to undergo viral amplification. Here, the virus must modulate the host innate immune response to prevent its elimination by the body while facilitating its replication. Replicated virus from the initially infected cell then disperse to infect neighbouring susceptible cells, possibly with spread to different cell types like leukocytes. This results in a localised infection, in which the virus mainly spreads and infects adjacent cells to the site of entry.[5][7] Otherwise, the virus can be released into extracellular fluids. Examples of localised infections include: common cold (rhinovirus), flu (parainfluenza), gastrointestinal infections (rotavirus) or skin infections (papillomavirus).[2]
Dissemination and secondary replication
In other cases, the virus can cause systemic disease through a disseminated infection spread throughout the body. The predominant mode of viral dissemination occurs through the blood or lymphatic system, some of which include viruses responsible for chickenpox (varicella zoster virus), smallpox (variola), HIV (human immunodeficiency virus). A minority of viruses can disseminate via the nervous system.[2][7] Notably, the poliovirus can be transmitted via the fecal-oral route, where it initially replicates in its site of entry, the small intestine and spread to regional lymph nodes. Then, the virus disseminates via the bloodstream into different organs in the body (e.g. liver, spleen), followed by a secondary round of replication and dissemination into the central nervous system to damage motor neurons.[4]
Shedding and secondary transmission
Finally, the viruses spread to sites where shedding into the environment can occur. The respiratory, alimentary and urogenital tracts and the blood are the most frequent sites of shedding in the form of bodily fluids, aerosols, skin, excrement. The virus would then go on to be transmitted to another person, and establish the infection cycle all over again.[2][4][7]
Factors affecting pathogenesis
There are a few main overarching factors affecting viral diseases:
- Virus tropism
- Virus factors
- Host factors
Molecular basis of virus tropism
Virus tropism refers to the virus' preferential site of replication in discrete cell types within an organ. In most cases, tropism is determined by the ability of the viral surface proteins to fuse or bind to surface receptors of specific target cells to establish infection. Thus, the binding specificity of viral surface proteins dictates tropism as well as the destruction of particular cell populations, and is therefore a major determinant of virus pathogenesis.[2][7] However, co-receptors are sometimes required in addition to the binding of cellular receptors on host cells to viral proteins in order to establish infection. For instance, HIV-1 requires target cells to express co-receptors CCR5 or CXCR4, on top of the CD4 receptor for productive viral attachment.[8] Interestingly, HIV-1 can undergo a tropism switch, where the virus glycoprotein gp120 initially uses CCR5 (mainly on macrophages) as the primary co-receptor for entering the host cell. Subsequently, HIV-1 switches to bind to CXCR4 (mainly on T cells) as the infection progresses, in doing so transitions the viral pathogenicity to a different stage.[8][9]
Apart from cellular receptors, viral tropism can also governed by other intracellular factors, such as tissue-specific transcription factors. An example would be the JC polyomavirus, in which its tropism is limited to glial cells since its enhancer is only active in glial cells,[2] and JC viral gene expression requires host transcription factors expressed exclusively in glial cells.[9]
The accessibility of host tissues and organs to the virus also regulates tropism. Accessibility is affected by physical barriers,[2][7] such as in enteroviruses, which replicate in the intestine since they are able to withstand bile, digestive enzymes and acidic environments.[9]
Virus factors
Viral genetics encoding viral factors will determine the degree of viral pathogenesis. This can be measured as virulence, which can be used to compare the quantitative degree of pathology between related viruses. In other words, different virus strains possessing different virus factors can lead to different degrees of virulence, which in turn can be exploited to study the differences in pathogenesis of viral variants with different virulence.[10][11]
Virus factors are largely influenced by viral genetics, which is the virulence determinant of structural or non-structural proteins and non-coding sequences. For a virus to successfully infect and cause disease in the host, it has to encode specific virus factors in its genome to overcome the preventive effects of physical barriers, and modulate host inhibition of virus replication.[2][10] In the case of poliovirus, all vaccine strains found in the oral polio vaccine contain attenuating point mutations in the 5' untranslated region (5' UTR). Conversely, the virulent strain responsible for causing polio disease does not contain these 5' UTR point mutations and thus display greater viral pathogenicity in hosts.[1][12]
Virus factors encoded in the genome often control the tropism, routes of virus entry, shedding and transmission. In polioviruses, the attenuating point mutations are thought to induce a replication and translation defect to reduce the virus' ability of cross-linking to host cells and replicate within the nervous system.[12]
Viruses have also developed a variety of immunomodulation mechanisms to subvert the host immune response. This tend to feature virus-encoded decoy receptors that target cytokines and chemokines produced as part of the host immune response, or homologues of host cytokines.[13][14] As such, viruses capable of manipulating the host cell response to infection as an immune evasion strategy exhibit greater pathogenicity.
Host factors
Viral pathogenesis is also largely dependent on host factors. Several viral infections have displayed a variety of effects, ranging from asymptomatic to symptomatic or even critical infection, solely based on differing host factors alone. In particular, genetic factors, age and immunocompetence play an important role is dictating whether the viral infection can be modulated by the host.[11][15] Mice that possess functional Mx genes encode an Mx1 protein which can selectively inhibit influenza replication. Therefore, mice carrying a non-functional Mx allele fail to synthesise the Mx protein and are more susceptible to influenza infection.[16] Alternatively, immunocompromised individuals due to existing illnesses may have a defective immune system which makes them more vulnerable to damage by the virus. Furthermore, a number of viruses display variable pathogenicity depending on the age of the host. Mumps, polio, and Epstein-Barr virus cause more severe disease in adults, while others like rotavirus cause more severe infection in infants. It is therefore hypothesized that the host immune system and defense mechanisms might differ with age.[10]
Disease mechanisms: How viral infections cause disease
A viral infection does not always cause disease. A viral infection simply involves viral replication in the host, but disease is the damage caused by viral multiplication.[5] An individual who has a viral infection but does not display disease symptoms is known as a carrier.[17]
Damage caused by the virus
Once inside host cells, viruses can destroy cells through a variety of mechanisms. Viruses often induce direct cytopathic effects to disrupt cellular functions.[11][18] This could be through releasing enzymes to degrade host metabolic precursors, or releasing proteins that inhibit the synthesis of important host factors, proteins, DNA and/or RNA.[13] Namely, viral proteins of herpes simplex virus can degrade host DNA and inhibit host cell DNA replication and mRNA transcription.[9] Poliovirus can inactivate proteins involved in host mRNA translation without affecting poliovirus mRNA translation. In some cases, expression of viral fusion proteins on the surface of the host cells can cause host cell fusion to form multinucleated cells. Notable examples include measles virus, HIV, respiratory syncytial virus.[2][13]
Importantly, viral infections can differ by the "lifestyle strategy". Persistent infections happen when cells continue to survive despite a viral infection and can be further classified into latent (only the viral genome is present, there is no replication occurring) and chronic (basal levels of viral replication without stimulating an immune response). In acute infections, lytic viruses are shed at high titres for rapid infection to a secondary tissue/host, whereas persistent viruses undergo shedding at lower titres for a longer duration of transmission (months to years).[1][2][19]
Lytic viruses are capable of destroying host cells by incurring and/or interfering with the specialised functions of host cells. An example would be the triggering of necrosis in host cells infected with the virus.[18] Otherwise, signatures of viral infection, like the binding of HIV to co-receptors CCR5 or CXCR4, can also trigger cell death via apoptosis through host signalling cascades by immune cells.[20] However, many viruses encode proteins that can modulate apoptosis depending on whether the infection is acute or persistent. Induction of apoptosis, such as through interaction with caspases, will promote viral shedding for lytic viruses to facilitate transmission, while viral inhibition of apoptosis could prolong the production of virus in cells, or allow the virus to remain hidden from the immune system in chronic, persistent infections.[9][11][18] Nevertheless, induction of apoptosis in major immune cells or antigen-presenting cells may also act as a mechanism of immunosuppression in persistent infections like HIV. The primary cause of immunosuppression in HIV patients is due to the depletion of CD4+ T helper cells.[4]
Interestingly, adenovirus has an E1A protein to induce apoptosis by initiating the cell cycle, and an E1B protein to block the apoptotic pathway through inhibition of caspase interaction.[21]
Persistent viruses can sometimes transform host cells into cancer cells.[15][22][18] Viruses such as the human papillomavirus (HPV), human T-lymphotropic virus (HTLV) etc., can stimulate growth of tumours in infected hosts, either by disrupting tumour suppressor gene expression (HPV) or upregulating proto-oncogene expression (HTLV).[15]
Damage caused by host immune system
Sometimes, instead of cell death or cellular dysfunction caused by the virus, the host immune response can mediate disease and excessive inflammation. The stimulation of the innate and adaptive immune system in response to viral infections destroys infected cells, which may lead to severe pathological consequences to the host. This damage caused by the immune system is known as virus-induced immunopathology.[23][24]
Specifically, immunopathology is caused by the excessive release of antibodies, interferons and pro-inflammatory cytokines, activation of the complement system, or hyperactivity of cytotoxic T cells. Secretion of interferons and other cytokines can trigger cell damage, fever and flu-like symptoms.[23][24] In severe cases of certain viral infections, as in avian H5N1 influenza in 2005, aberrant induction of the host immune response can elicit a flaring release of cytokines known as a cytokine storm.[25]
In some instances, viral infection can initiate an autoimmune response, which occurs via different proposed mechanisms: molecular mimicry and bystander mechanism.[26] Molecular mimicry refers to an overlap in structural similarity between a viral antigen and a self-antigen.[26] The bystander mechanism hypothesizes the initiation of a non-specific and overreactive antiviral response that tackles self-antigens in the process.[26] Damage caused by the host itself due to autoimmunity was observed in the West Nile virus.[27]
Incubation Period
Viruses display variable incubation periods upon virus entry into the host. The incubation period refers to the time taken for the onset of disease after first contact with the virus.[2][7] In Rabiesvirus, the incubation period varies with the distance traversed by the virus to the target organ; but in most viruses the length of incubation depends on many factors.[7][28] Surprisingly, generalised infections by togaviruses have a short incubation period due to the direct entry of the virus into target cells through insect bites.[7]
There are several other factors that affect the incubation period. The mechanisms behind long incubation periods, months or years for example, are not completely understood yet.[28]
Evolution of virulence
Some relatively avirulent viruses in their natural host show increased virulence upon transfer to a new host species. When an emerging virus first invades a new host species, the hosts have little or no immunity against the virus and often experience high mortality. Over time, a decrease in virulence in the predominant strain can sometimes be observed. A successful pathogen needs to spread to at least one other host, and lower virulence can result in higher transmission rates under some circumstances. Likewise, genetic resistance against the virus can develop in a host population over time.[2][29]
An example of the evolution of virulence in emerging virus is the case of myxomatosis in rabbits. The release of wild European rabbits in 1859 into Victoria, Australia for sport resulted in a rabbit plague. In order to curb with rabbit overpopulation, myxoma virus, a lethal species-specific poxvirus responsible for myxomatosis in rabbits, was deliberately released in South Australia in 1950. This led to a 90% decrease in rabbit populations, and the disease became endemic in a span of five years. Significantly, severely attenuated strains of the myxoma virus were detected in merely 2 years of its release, and genetic resistance in rabbits emerged within seven years.[30]
See also
- Virology
- Glossary of virology
- Pathogen
- Pathogenesis
- List of human diseases associated with infectious pathogens
References
- 1 2 3 Nathanson N (2016-01-04). Viral pathogenesis. Lippincott-Raven. pp. 2016. ISBN 9780128011744.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 Albrecht T, Fons M, Boldogh I, Rabson AS (1996-01-01). Baron S (ed.). Medical Microbiology (4th ed.). Galveston (TX): University of Texas Medical Branch at Galveston. ISBN 0963117211. PMID 21413282. Archived from the original on 2019-12-16. Retrieved 2022-06-20.
- 1 2 Racaniello V. "Viral Pathogenesis" (PDF). Archived (PDF) from the original on 6 October 2021. Retrieved 8 February 2014.
- 1 2 3 4 5 Ryan KJ, Ray CG, eds. (2014). "Chapter 7 Viral Pathogenesis". Sherris Medical Microbiology (6 ed.).
- 1 2 3 4 Morse SA, Riedel S, Mietzner TA, Miller S (2019-08-25). Jawetz Melnick & Adelbergs Medical Microbiology 28E. McGraw-Hill Education. ISBN 9781260012033.
- ↑ Chapter 33 (Disease summaries), pp. 367–92 in:Fisher, Bruce; Harvey, Richard P.; Champe, Pamela C. (2007). Lippincott's Illustrated Reviews: Microbiology. Lippincott's Illustrated Reviews Series. Hagerstwon, MD: Lippincott Williams & Wilkins. pp. 367–92. ISBN 978-0-7817-8215-9.
- 1 2 3 4 5 6 7 8 9 Mitchell MG (2010-04-16). Molecular Pathology and the Dynamics of Disease. Academic Press. doi:10.1016/C2016-0-04893-3. ISBN 978-0-12-814610-1.
- 1 2 Shen HS, Yin J, Leng F, Teng RF, Xu C, Xia XY, Pan XM (February 2016). "HIV coreceptor tropism determination and mutational pattern identification". Scientific Reports. 6: 21280. Bibcode:2016NatSR...621280S. doi:10.1038/srep21280. PMC 4756667. PMID 26883082.
- 1 2 3 4 5 Kumar V, Abbas AK, Aster JC, eds. (2014). Robbins & Cotran Pathologic Basis of Disease (9 ed.). Elsevier. ISBN 9780323313094.
- 1 2 3 Cann A (2016-01-04). Jawetz, Melnick, & Adelberg's Medical Microbiology (28 ed.). Elsevier. doi:10.1016/B978-0-12-801946-7.00007-9. ISBN 9780128011744. S2CID 215745990.
- 1 2 3 4 Fuentes-González AM, Contreras-Paredes A, Manzo-Merino J, Lizano M (June 2013). "The modulation of apoptosis by oncogenic viruses". Virology Journal. 10: 182. doi:10.1186/1743-422X-10-182. PMC 3691765. PMID 23741982.
- 1 2 Gutiérrez AL, Denova-Ocampo M, Racaniello VR, del Angel RM (May 1997). "Attenuating mutations in the poliovirus 5' untranslated region alter its interaction with polypyrimidine tract-binding protein". Journal of Virology. 71 (5): 3826–33. doi:10.1128/JVI.71.5.3826-3833.1997. PMC 191533. PMID 9094658.
- 1 2 3 MacLachlan J, Dubovi E (2011). Fenner's Veterinary Virology (4th ed.). Elsevier. ISBN 9780123751584.
- ↑ Felix J, Savvides SN (February 2017). "Mechanisms of immunomodulation by mammalian and viral decoy receptors: insights from structures". Nature Reviews. Immunology. 17 (2): 112–129. doi:10.1038/nri.2016.134. PMID 28028310. S2CID 4058941.
- 1 2 3 Dimmock NJ, Easton AJ, Leppard KN, eds. (2016). Introduction to modern virology (7 ed.). John Wiley & Sons Ltd. ISBN 9781119978107.
- ↑ Staeheli P, Grob R, Meier E, Sutcliffe JG, Haller O (October 1988). "Influenza virus-susceptible mice carry Mx genes with a large deletion or a nonsense mutation". Molecular and Cellular Biology. 8 (10): 4518–23. doi:10.1128/mcb.8.10.4518. PMC 365527. PMID 2903437.
- ↑ Furuya-Kanamori L, Cox M, Milinovich GJ, Magalhaes RJ, Mackay IM, Yakob L (June 2016). "Heterogeneous and Dynamic Prevalence of Asymptomatic Influenza Virus Infections". Emerging Infectious Diseases. 22 (6): 1052–6. doi:10.3201/eid2206.151080. PMC 4880086. PMID 27191967.
- 1 2 3 4 Cann A (2015). Principles of Molecular Virology (6 ed.). Academic Press. ISBN 9780128019559.
- ↑ Flint SJ, Racaniello VR, Rall GF, Skalka AM, Enquist LW (2015). Principles of Virology, 4th Edition (4th ed.). ASM Press. ISBN 978-1-555-81933-0.
- ↑ Ahr B, Robert-Hebmann V, Devaux C, Biard-Piechaczyk M (June 2004). "Apoptosis of uninfected cells induced by HIV envelope glycoproteins". Retrovirology. 1: 12. doi:10.1186/1742-4690-1-12. PMC 446229. PMID 15214962. S2CID 18931635.
- ↑ White E (1998). "Regulation of Apoptosis by Adenovirus E1A and E1B Oncogenes". Seminars in Virology. 8 (6): 505–513. doi:10.1006/smvy.1998.0155.
- ↑ Mothes W, Sherer NM, Jin J, Zhong P (September 2010). "Virus cell-to-cell transmission". Journal of Virology. 84 (17): 8360–8. doi:10.1128/JVI.00443-10. PMC 2918988. PMID 20375157.
- 1 2 Rouse BT (1996). "Virus-induced immunopathology". Advances in Virus Research. 47: 353–76. doi:10.1016/S0065-3527(08)60739-3. ISBN 9780120398478. PMC 7130923. PMID 8895836.
- 1 2 Rouse BT, Sehrawat S (July 2010). "Immunity and immunopathology to viruses: what decides the outcome?". Nature Reviews. Immunology. 10 (7): 514–26. doi:10.1038/nri2802. PMC 3899649. PMID 20577268.
- ↑ Tisoncik JR, Korth MJ, Simmons CP, Farrar J, Martin TR, Katze MG (March 2012). "Into the eye of the cytokine storm". Microbiology and Molecular Biology Reviews. 76 (1): 16–32. doi:10.1128/MMBR.05015-11. PMC 3294426. PMID 22390970.
- 1 2 3 Smatti MK, Cyprian FS, Nasrallah GK, Al Thani AA, Almishal RO, Yassine HM (August 2019). "Viruses and Autoimmunity: A Review on the Potential Interaction and Molecular Mechanisms". Viruses. 11 (8): 762. doi:10.3390/v11080762. PMC 6723519. PMID 31430946.
- ↑ Hawkes MA, Hocker SE, Leis AA (December 2018). "West Nile virus induces a post-infectious pro-inflammatory state that explains transformation of stable ocular myasthenia gravis to myasthenic crises". Journal of the Neurological Sciences. 395: 1–3. doi:10.1016/j.jns.2018.09.015. PMID 30267806. S2CID 52894428.
- 1 2 Nelson KE, Williams CM (2013). Infectious Disease Epidemiology: Theory and Practice (3 ed.). Jones & Bartlett Learning. ISBN 978-1-44-968379-5.
- ↑ Bolker BM, Nanda A, Shah D (May 2010). "Transient virulence of emerging pathogens". Journal of the Royal Society, Interface. 7 (46): 811–22. doi:10.1098/rsif.2009.0384. PMC 2874237. PMID 19864267.
- ↑ Kerr PJ (March 2012). "Myxomatosis in Australia and Europe: a model for emerging infectious diseases". Antiviral Research. 93 (3): 387–415. doi:10.1016/j.antiviral.2012.01.009. PMID 22333483.