X-linked reticulate pigmentary disorder
| X-linked reticulate pigmentary disorder | |
|---|---|
| Other names: Familial cutaneous amyloidosis,[1] Partington amyloidosis,[1] Partington cutaneous amyloidosis,[1] Partington syndrome type II,[1] reticulate pigmentary disorder,[1] X-linked reticulate pigmentary disorder with systemic manifestations[1] | |
.svg.png.webp) | |
| This condition is inherited in an X-linked recessive manner. Carrier females usually only have linear streaks of hyperpigmentation. | |
X-linked reticulate pigmentary disorder is a rare X-linked genetic condition in which males manifest multiple systemic symptoms and a reticulated mottled brown pigmentation of the skin, which, on biopsy, demonstrated dermal deposits of amyloid. Females usually only have linear streaks of hyperpigmentation.[1]
The syndrome is also referred to by the acronym X-Linked-PDR or XLPRD.[2] It's a very rare disease, genetically determined, with a chronic course.
It was characterized in 1981.[3]
Signs and symptoms
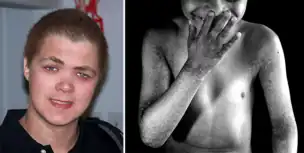
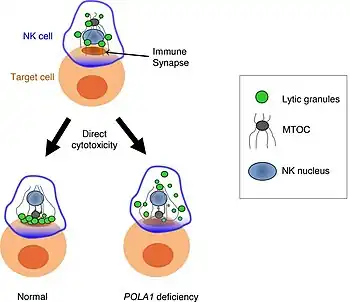
Affected males develop generalized reticular hyperpigmentation in early childhood. Hair often looks bedraggled or brushed backward, hanging low on the forehead. Under XLPDR conditions, autoimmune manifestations are developed due to chronically activated anti-viral type I interferon response, connecting XLPDR with disorders like Aicardi-Goutiere syndrome, Systemic Lupus Erythematosus, Psoriasis, etc. 3 Archived 2023-02-22 at the Wayback Machine Meanwhile, another typical symptom - immunodeficiency - can be developed due to discovering a functional defect in the cytolytic activity of NK cells. Starokadomskyy at al. discovered that POLA1 deficiency is associated with decreased direct cytotoxicity of NK cells due to disturbances in vesicular traffic. Meanwhile, antibody-dependent cell cytotoxicity (ADCC) remains unchanged in XLPDR NK cells.[4]
The most common manifestations of XLPDR:
- Recurrent respiratory infections
- Dyskeratosis corneal
- Photophobia
- Hypohidrosis (lack of sweat glands)
- NK cell functional deficiency
- Growth retardation
- Gastrointestinal disorders
- Kidney disease
- Kidney stones
- Urinary infections
- Webbed feet or hands
- Electrolyte imbalance
- Retinitis pigmentosa
- Lymphoedema
- Thyroid abnormalities
Not every patient shows all of the listed symptoms. However, skin pathologies, recurrent lung infection, high titer of interferon type I in the blood, and impaired direct cytotoxicity of NK cells are the most common symptoms. In females the disease is characterized by skin rashes linear hyper pigmentation following the Blaschko's lines, morphologically similar to stage 3 pigment incontinence. There are no systemic manifestations associated with XLPDR in females.
Most XLPDR patients stabilize with age and have an overall less complicated clinical course after adolescence. Gastrointestinal and urinary tract complications are progressively less active, and the pace of infections tends to decrease. However, those who have severe lung damage remain prone to recurrent pneumonia and may succumb to severe infections. Hypohidrosis is irreversible and remains a problem for life. XLPDR patients have normal fertility and the mutation has been transmitted to their female offspring.[2][5]
Mechanism
Mutation of the POLA1 gene leads to loss of expression of the catalytic subunit of DNA polymerase-α and is responsible for XLPDR.[2]
Loss of POLA1 expression results in reduced levels of RNA:DNA hybrids in the cytosol and unexpectedly triggers aberrant immune responses (e.g. type I interferon production) which at least in part can account for the symptoms associated with XLPDR.[2]
Another trigger of the immunodeficiency phenotype is a functional deficiency of NK cells, major players of innate antiviral immune system.[4]
Diagnosis
All XLPDR probands shared the same unique intronic variant mapping to intron 13 of POLA1, (NM_016937.3:c.1375-354A>G). XLPDR lacks allelic heterogeneity, meaning that the disorder is uniquely associated with the NM_016937.3:c.1375-354A>G intronic variant.[2][5] The final diagnosis usually requires PCR or WGS confirmation.
Treatment
Management of other XLPDR symptoms is largely supportive. Conventional management of recurrent lung infections with antibiotics is essential; many patients receive inhaled prophylactic management akin to cystic fibrosis patients. Urethral strictures are treated with sequential dilations. Eye involvement is progressive, leading to blindness, and recurs after corneal transplantation.
Recently, a number of reports suggest encouraging results with the use of JAK inhibitors baricitinib and ruxolitinib in several distinct type I interferonopathies. In fact, one XLPDR patient with refractory colitis was treated with tofacitinib with positive response of the colitis and no exacerbation of pulmonary infections.[5]
Other options that may be worth considering in the future are interferon receptor neutralizing antibodies, which are being actively pursued in the treatment of lupus where they show particular promise. A path for definitive treatment for XLPDR is at present unclear, but it is tempting to speculate whether the immunologic disturbance is predominantly driven by the hematopoietic compartment. The clinical course of the eye involvement is consistent with this possibility. If so, the disorder might be amenable to hematopoietic stem cell transplantation and could even be suitable for gene therapy and autologous stem cell transplant.
See also
References
- 1 2 3 4 5 6 7 Rapini RP, Bolognia JL, Jorizzo JL (2007). Dermatology: 2-Volume Set. St. Louis: Mosby. p. 630. ISBN 978-1-4160-2999-1.
- 1 2 3 4 5 Starokadomskyy P, Gemelli T, Rios JJ, Xing C, Wang RC, Li H, et al. (May 2016). "DNA polymerase-α regulates the activation of type I interferons through cytosolic RNA:DNA synthesis". Nature Immunology. 17 (5): 495–504. doi:10.1038/ni.3409. PMC 4836962. PMID 27019227.
- ↑ Partington MW, Marriott PJ, Prentice RS, Cavaglia A, Simpson NE (1981). "Familial cutaneous amyloidosis with systemic manifestations in males". American Journal of Medical Genetics. 10 (1): 65–75. doi:10.1002/ajmg.1320100109. PMID 6794369.
- 1 2 Starokadomskyy P, Wilton KM, Krzewski K, Lopez A, Sifuentes-Dominguez L, Overlee B, et al. (November 2019). "NK cell defects in X-linked pigmentary reticulate disorder". JCI Insight. 4 (21). doi:10.1172/jci.insight.125688. PMC 6948767. PMID 31672938.
- 1 2 3 Starokadomskyy P, Escala Perez-Reyes A, Burstein E (February 2021). "Immune Dysfunction in Mendelian Disorders of POLA1 Deficiency". Journal of Clinical Immunology. 41 (2): 285–293. doi:10.1007/s10875-020-00953-w. ISSN 0271-9142. PMC 7864891. PMID 33392852.
External links
| Classification | |
|---|---|
| External resources |
|