ZTTK syndrome
| ZTTK syndrome | |
|---|---|
| Other names | Zhu-Tokita-Takenouchi-Kim syndrome |
ZTTK Syndrome (Zhu-Tokita-Takenouchi-Kim syndrome) is a rare disease caused in humans by a genetic mutation of the SON gene. Common symptoms include developmental delay and sometimes moderate to several intellectual disability.[1][2]
Characteristic abnormalities include cerebral cortex malformations, vision difficulties, musculoskeletal abnormalities and congenital defects.[1] Individuals with a mutation in the SON gene may not all display these features. However, SON loss of function (LoF) variants appear to cause a clinically distinguished phenotype.[1]
Signs and Symptoms
The key signs and symptoms associated with ZTTK Syndrome patients include ocular, facial and systemic features.
Ocular Features
Distinctive ocular features of the ZTTK syndrome are deep-set eyes, down-slanting palpebral fissures and horizontal eyebrows.[1] Children with ZTTK syndrome may present with vision problems including optic atrophy and cerebral visual impairment, resulting in poor visual responses.[1] Strabismus; misalignment or crossing of the eyes when viewing an object, direct hypermetropia; farsightedness, and nystagmus; eyes making repetitive and uncontrolled movements, are frequently present.[3]
Facial Features
Individuals with ZTTK syndrome have distinctive minor to moderate facial dysmorphisms. Distinct facial features include facial asymmetry, low-set ears, midface retraction, frontal bossing,[4] a depressed and or broad nasal bridge and a smooth or short philtrum.[1]
Systemic Features
Multi-system abnormalities are common in ZTTK syndrome. The majority of individuals diagnosed with ZTTK syndrome display congenital malformations such as urogenital and malformations, heart defects, and a high or cleft palate.[1]
Congenital defects such as a thinned atrial septum, ventricular septal defects, patent ductus arteriosus, dysplastic kidney and agenesis of the lung and gallbladder have also been noted.[4] Whole body musculoskeletal abnormalities have been observed in ZTTK syndrome patients, including hemivertebrae, scoliosis or kyphosis, contractures, joint laxity,[4] joint hypermobility and hypotonia.[1] During the neonatal period, persistent feeding difficulties is associated with growth failure and a short stature in most individuals with ZTTK syndrome.[4]
Central Nervous System
Developmental delay is common in ZTTK syndrome patients, and appears to progressively increase the severity of intellectual disability with age.[1] The development of gross and fine motor skills, as well as fluent and receptive language skills are shown to be delayed in developmental age. Macrocephaly and brain white matter abnormalities have also been observed.[5] Seizures often develop between the ages of 1 to 6 years old.[3]
Physiological
Mutations of the SON gene can affect metabolism and mitochondrial function in newborns with ZTTK syndrome. Metabolic screening confirmed mitochondrial dysfunction and O-glycosylation defects in individuals with ZTTK syndrome.[1] Decreased levels of immunoglobulin A and or immunoglobulin G identified in ZTTK syndrome patients resulted in coagulation abnormalities.[2]
Genetics
ZTTK syndrome is caused by heterozygous mutations in the SON gene.[5] As an autosomal dominant disease, children with parents carrying a SON mutation have a 50% risk of inheriting the mutation. However, the majority of affected individuals have de novo mutations in the SON gene and ZTTK syndrome is not inherited to their children.[3]
Allelic Variants of SON Gene
Many individuals with ZTTK syndrome have identified heterozygosity for a de novo 4-base pair deletion[5][6], de novo mutation in exon 3 in the SON gene[1] and de novo 2-base point insertion in exon,[1] resulting in haploinsufficiency or a frameshift and premature termination in the arginine/serine (RS) domain. Peripheral blood cells from the sampled patients confirmed decreased levels of the mutant RNA transcript, consistent with haploinsufficiency.[1] Other mutations observed include a nonsense mutation, an in-frame deletion of amino acids and an entire gene deletion.[1] De novo heterozygous 1-base point duplication in exon 3 and 1-base point deletion in exon 4 of the SON gene resulted in a frameshift and premature termination.[4] Parental DNA has confirmed that de novo mutations are common in patients with ZTTK syndrome.[1] De novo LoF mutations and haploinsufficiency for the SON gene are shown to cause profound developmental malformations during embryonic development as seen in the phenotypic manifestations of the ZTTK syndrome.[4]
Structure of SON Gene
SON is a large protein consisting of 2426 amino acids and repeat sequences.[7] SON is located within the human chromosomal region 21q22.11 in nuclear speckles and consists of 12 exons.[8] Exon 3 of the SON gene is particularly large, accounting for 82% of the entire coding region.[1] The majority of SON variants found in ZTTK syndrome individuals are localised to exon 3.[4]
Mechanism
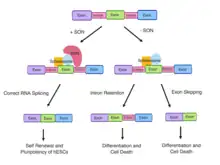
Role of SON in RNA Splicing
The SON gene encodes the SON protein, which is able to bind to DNA and RNA.[9] The SON protein is mainly localised to nuclear speckles and involved in a variety of cellular processes such as transcription, cell cycle regulation and subnuclear organisation of pre-messenger RNA (mRNA) splicing.[9][10]
SON contains various domains such as the RS-rich domain, a G-patch domain and a double-stranded RNA-binding motif.[7][11] The presence of these domains is necessary for SON to mediate constitutive and alternative splicing.[1] The RS-rich domain serves to localise SON in nuclear speckles with pre-mRNA processing factors.[9] The functional domains and specific localisation of SON in nuclear speckles has indicated its role in pre-mRNA splicing.[9]
SON also plays a key role in alternative splicing of exons. SON is required for genome stability by ensuring the efficiency of RNA splicing of weak constitutive and alternative splice sites. SON-dependent cell-cycle genes possess a weak 5’ or 3’ splice site and are dependent on SON to ensure efficient splicing and spliceosome recognition.[7]
Role of SON in Embryonic Development
The SON gene also plays a critical role during development. SON is expressed preferentially in undifferentiated stem cells.[9] Depletion of SON results in stem cell differentiation.[9]
Human embryonic stem cells (hESCs) are able to undergo lineage-specific differentiation into specific types of cells, known as pluripotency.[12] Pluripotent stem cells, such as hESCs can undergo gastrulation to give rise to the three germ layers.[9]
A significant level of SON expression in fetal tissue has suggested a regulatory role of SON in cellular proliferation and or differentiation during embryonic development by influencing the splicing of pluripotency maintenance genes.[13] The expression of transcription factors such as the SON factor and epigenetic modifiers regulate the pluripotency of hESCs by ensuring genes undergo RNA splicing to create a mature RNA transcript.[14]
The SON gene is required for RNA splicing of transcripts encoding the cell-cycle protein TUBG1 and genes maintaining hESC pluripotency; PRDM14, OCTA, E4F1 and MED24 in hESCs.[12] As OCT4 is involved in the core transcriptional circuitry in hESCs, misregulation of OCT4 induces cell differentiation. PRDM14 is a pluripotency regulator and MED24 is a mediator complex essential in the maintenance of pluripotency.[12] In wild-type ESCs, SON binding to the RNA transcripts of pluripotency regulating genes such as PRDM14 and OCT4 results in correct splicing and maintenance of pluripotency.[14]
Effects of SON Haploinsufficiency on RNA Splicing and Embryonic Development
The downregulation of SON can impact the regulation of mitotic regulator transcripts and cause defects in cell survival and the developmental process.[9] SON depletion causes decreased cell growth,[7][15][16] disarrayed microtubule processes and disordered spindle pole separation, causing mitotic arrest at metaphase and severe genome integrity impairment.[7][15][16] Mitotic cells without functional SON have increased double-stranded DNA breaks and micronuclei formation.[15] Consequently, genome stability and regulation of the cell cycle are compromised, contributing to the development of multi-organ defects in ZTTK syndrome patients.[7]
Aberrant splicing and de novo heterozygous LoF mutations in SON gene disrupts the process of gene expression and can result in SON haploinsufficiency.[17][5] ZTTK syndrome individuals with SON haploinsufficiency display decreased mRNA expression and abnormal RNA splicing products of numerous genes which are necessary for neuronal cell migration, metabolic processes and neurodevelopment of the brain.[5]
RNA analyses from affected individuals with ZTTK syndrome confirmed the downregulation of genes essential for neuronal migration and cortex organisation (TUBG1, FLNA, PNKP, WDR62, PSMD3, HDAC6) and metabolism (PCK2, PFKL, IDH2, ACY1, and ADA).[1] Aberrant SON-mediated RNA splicing results from the accumulation of mis-spliced transcripts.[1] The mis-spliced RNA products are caused by significant intron retention (TUBG1, FLNA, PNKP, WDR62, PSMD3, PCK2, PFKL, IDH2, and ACY1) and exon skipping (HDAC6 and ADA).[1] In contrast, the parents of individuals with ZTTK syndrome display an absence of mis-spliced RNA products.[1]
SON depletion downregulates and causes aberrant splicing of the pluripotency factors, OCT4, PRDM14, MED24 and E4F1, inducing spontaneous differentiation of hESCs followed by widespread cell death.[12][14] As SON acts as an intron splicing activator, the depletion of SON leads to increased intron retention and exon skipping in hESCs in regulatory genes of the cell cycle and hESC identity.[18] Mutations in the SON gene and or SON haploinsufficiency compromises SON-mediated RNA splicing and contributes to the complex developmental defects observed in individuals with ZTTK syndrome.[1] Erroneous SON function causes insufficient production of downstream targets, genome instability and disrupted cell cycle progression which are fundamental to the developmental defects and organ abnormalities in individuals with ZTTK syndrome. For example, FLNA haploinsufficiency observed in individuals with ZTTK syndrome is the main cause of a rare brain disorder, periventricular nodular heterotopia. De novo LoF mutations in TUBG1 can result in microcephaly and cortical malformations due to compromised SON-mediated RNA splicing in affected ZTTK syndrome individuals.[19]
The consequence of SON haploinsufficiency on embryonic development has also been studied in zebrafish animal models (Danio rerio). A range of developmental defects was observed, including bent, shortened or gnarled tails, massive body curvatures with deformed body axes, eye malformations and microcephaly.[1] Embryos that survived for a longer period of time have more severe phenotypes such as spinal malformations with brain oedema, imitating features observed in affected ZTTK syndrome individuals.[1]
Diagnosis
Brain Imaging
Early diagnosis of the ZTTK syndrome can be determined by brain imaging. Magnetic resonance imaging (MRI) of the brain of ZTTK syndrome patients have revealed significant abnormalities.[1]
Abnormal gyration patterns were seen, including polymicrogyria; many unusually small folds in the brain, simplified gyria; reduced number and shallow appearance of gyri, and periventricular nodular heterotopia; failure of neurons to migrate properly during early development of the fetal brain.[3][20]
Ventriculomegaly can also be observed in MRI where the lateral ventricles become dilated in the foetus and can contribute to developmental delays in ZTTK syndrome individual.[3] Another common feature observed in ZTTK syndrome patients is Arnold-Chiari malformations which are structural defects in the cerebellum that manifest during fetal development and can lead to vision problems, scoliosis or kyphosis in ZTTK syndrome patients.[21]
Other pathological features seen on MRI scans of ZTTK syndrome individuals include arachnoid cysts, hypoplasia of the corpus callosum and cerebellar hemispheres and loss of periventricular white matter.[1]
Most individuals with ZTTK syndrome are identified early in childhood due to developmental delays and intellectual disabilities.[22] However, a formal diagnosis of intellectual disability can only be conducted by a performance of an IQ test score of below 70.[21]
Whole Exome Sequencing
Whole exome sequencing (WES) can be used as a non-biased tool in the diagnostic evaluation of individuals with suspected genetic disorders such as the ZTTK syndrome.[1] Using WES, individuals were identified with truncating variants of SON and overlapping clinical features.
ZTTK syndrome has been identified as a neurodevelopmental disorder associated with a de novo mutation in the SON gene using WES. The SON gene is known to be a major cause of severe intellectual disability and consequent developmental disorders.[22] The first de novo truncating variant in SON was recognised in a group of individuals with severe intellectual disabilities.[5] Sanger sequencing or the use of WES of parental samples confirmed the de novo status of the truncating and missense mutations of the SON gene in the sampled ZTTK syndrome individuals.[1] Variants identified included a premature stop variant in exon 3, frame-shift variants in exon 3 and a frameshift variant in exon 4.[1]
Treatment
There is currently no treatment for ZTTK syndrome. However, physical therapy and addressing the specific problems of multi organ disorders may be helpful.[3] The main focus should be on the diagnosis and care of individuals with ZTTK syndrome.
References
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 Kim, Jung-Hyun; Shinde, Deepali N; Reijnders, Margot R.F; Hauser, Natalie S; Belmonte, Rebecca L; Wilson, Gregory R; Bosch, Daniëlle G.M; Bubulya, Paula A; Shashi, Vandana; Petrovski, Slavé; Stone, Joshua K; Park, Eun Young; Veltman, Joris A; Sinnema, Margje; Stumpel, Connie T.R.M; Draaisma, Jos M; Nicolai, Joost; Yntema, Helger G; Lindstrom, Kristin; De Vries, Bert B.A; Jewett, Tamison; Santoro, Stephanie L; Vogt, Julie; Bachman, Kristine K; Seeley, Andrea H; Krokosky, Alyson; Turner, Clesson; Rohena, Luis; Hempel, Maja; Kortüm, Fanny; et al. (2016). "De Novo Mutations in SON Disrupt RNA Splicing of Genes Essential for Brain Development and Metabolism, Causing an Intellectual-Disability Syndrome". The American Journal of Human Genetics. 99 (3): 711–719. doi:10.1016/j.ajhg.2016.06.029. PMC 5011044. PMID 27545680.
- 1 2 "OMIM Entry # 617140 - ZTTK SYNDROME; ZTTKS". Online Mendelian Inheritance in Man. Johns Hopkins University. Retrieved 27 October 2017.
- 1 2 3 4 5 6 "ZTTK Syndrome".
- 1 2 3 4 5 6 7 Tokita, Mari J.; Braxton, Alicia A.; Shao, Yunru; Lewis, Andrea M.; Vincent, Marie; Küry, Sébastien; Besnard, Thomas; Isidor, Bertrand; Latypova, Xénia (September 2016). "De Novo Truncating Variants in SON Cause Intellectual Disability, Congenital Malformations, and Failure to Thrive". The American Journal of Human Genetics. 99 (3): 720–727. doi:10.1016/j.ajhg.2016.06.035. ISSN 0002-9297. PMC 5011061. PMID 27545676.
- 1 2 3 4 5 6 Zhu, Xiaolin; Petrovski, Slavé; Xie, Pingxing; Ruzzo, Elizabeth K.; Lu, Yi-Fan; McSweeney, K. Melodi; Ben-Zeev, Bruria; Nissenkorn, Andreea; Anikster, Yair (2015-01-15). "Whole-exome sequencing in undiagnosed genetic diseases: interpreting 119 trios". Genetics in Medicine. 17 (10): 774–781. doi:10.1038/gim.2014.191. ISSN 1098-3600. PMC 4791490. PMID 25590979.
- ↑ Takenouchi, Toshiki; Miura, Kiyokuni; Uehara, Tomoko; Mizuno, Seiji; Kosaki, Kenjiro (2016-06-03). "EstablishingSONin 21q22.11 as a cause a new syndromic form of intellectual disability: Possible contribution to Braddock-Carey syndrome phenotype". American Journal of Medical Genetics Part A. 170 (10): 2587–2590. doi:10.1002/ajmg.a.37761. ISSN 1552-4825. PMID 27256762.
- 1 2 3 4 5 6 Ahn, Eun-Young; DeKelver, Russell C.; Lo, Miao-Chia; Nguyen, Tuyet Ann; Matsuura, Shinobu; Boyapati, Anita; Pandit, Shatakshi; Fu, Xiang-Dong; Zhang, Dong-Er (April 2011). "SON Controls Cell-Cycle Progression by Coordinated Regulation of RNA Splicing". Molecular Cell. 42 (2): 185–198. doi:10.1016/j.molcel.2011.03.014. ISSN 1097-2765. PMC 3137374. PMID 21504830.
- ↑ Khan, I. M.; Fisher, R. A.; Johnson, K. J.; Bailey, M. E. S.; Siciliano, M. J.; Kessling, A. M.; Farrer, M.; Carritt, B.; Kamalati, T. (January 1994). "The SON gene encodes a conserved DNA binding protein mapping to human chromosome 21". Annals of Human Genetics. 58 (1): 25–34. doi:10.1111/j.1469-1809.1994.tb00723.x. ISSN 0003-4800. PMID 8031013. S2CID 31519119.
- 1 2 3 4 5 6 7 8 Lu, Xinyi; Ng, Huck-Hui; Bubulya, Paula A. (2014-04-30). "The role of SON in splicing, development, and disease". Wiley Interdisciplinary Reviews: RNA. 5 (5): 637–646. doi:10.1002/wrna.1235. ISSN 1757-7004. PMC 4138235. PMID 24789761.
- ↑ Spector, D. L.; Lamond, A. I. (2010-10-06). "Nuclear Speckles". Cold Spring Harbor Perspectives in Biology. 3 (2): a000646. doi:10.1101/cshperspect.a000646. ISSN 1943-0264. PMC 3039535. PMID 20926517.
- ↑ Hickey, Christopher J.; Kim, Jung-Hyun; Ahn, Eun-Young Erin (2013-12-13). "New Discoveries of Old SON: A Link Between RNA Splicing and Cancer". Journal of Cellular Biochemistry. 115 (2): 224–231. doi:10.1002/jcb.24672. ISSN 0730-2312. PMID 24030980. S2CID 23130360.
- 1 2 3 4 Lu, Xinyi; Göke, Jonathan; Sachs, Friedrich; Jacques, Pierre-Étienne; Liang, Hongqing; Feng, Bo; Bourque, Guillaume; Bubulya, Paula A.; Ng, Huck-Hui (2013-09-08). "SON connects the splicing-regulatory network with pluripotency in human embryonic stem cells". Nature Cell Biology. 15 (10): 1141–1152. doi:10.1038/ncb2839. ISSN 1465-7392. PMC 4097007. PMID 24013217.
- ↑ Cheng, Suzanne; Lutfalla, Georges; Uze, Gilles; Chumakov, Ilya M.; Gardiner, Katheleen (1993). "GART, SON, IFNAR, and CRF2-4 genes cluster on human Chromosome 21 and mouse Chromosome 16". Mammalian Genome. 4 (6): 338–342. doi:10.1007/bf00357094. ISSN 0938-8990. PMID 8318737. S2CID 19770065.
- 1 2 3 Livyatan, Ilana; Meshorer, Eran (October 2013). "SON sheds light on RNA splicing and pluripotency". Nature Cell Biology. 15 (10): 1139–1140. doi:10.1038/ncb2851. ISSN 1465-7392. PMID 24084863. S2CID 12137904.
- 1 2 3 Huen, Michael S.Y.; Sy, Shirley M.H.; Leung, Ka Man; Ching, Yick-Pang; Tipoe, George L.; Man, Cornelia; Dong, Shuo; Chen, Junjie (July 2010). "SON is a spliceosome-associated factor required for mitotic progression". Cell Cycle. 9 (13): 2679–2685. doi:10.4161/cc.9.13.12151. ISSN 1538-4101. PMC 3040851. PMID 20581448.
- 1 2 Sharma, Alok; Takata, Hideaki; Shibahara, Kei-ichi; Bubulya, Athanasios; Bubulya, Paula A. (2010-02-15). "Son Is Essential for Nuclear Speckle Organization and Cell Cycle Progression". Molecular Biology of the Cell. 21 (4): 650–663. doi:10.1091/mbc.e09-02-0126. ISSN 1059-1524. PMC 2820428. PMID 20053686.
- ↑ Cooper, Thomas A.; Wan, Lili; Dreyfuss, Gideon (February 2009). "RNA and Disease". Cell. 136 (4): 777–793. doi:10.1016/j.cell.2009.02.011. ISSN 0092-8674. PMC 2866189. PMID 19239895.
- ↑ Juan-Mateu, Jonàs; Villate, Olatz; Eizirik, Décio L (May 2016). "MECHANISMS IN ENDOCRINOLOGY: Alternative splicing: the new frontier in diabetes research". European Journal of Endocrinology. 174 (5): R225–R238. doi:10.1530/eje-15-0916. ISSN 0804-4643. PMC 5331159. PMID 26628584.
- ↑ Poirier, Karine; Lebrun, Nicolas; Broix, Loic; Tian, Guoling; Saillour, Yoann; Boscheron, Cécile; Parrini, Elena; Valence, Stephanie; Pierre, Benjamin Saint (2013-04-21). "Mutations in TUBG1, DYNC1H1, KIF5C and KIF2A cause malformations of cortical development and microcephaly". Nature Genetics. 45 (6): 639–647. doi:10.1038/ng.2613. ISSN 1061-4036. PMC 3826256. PMID 23603762.
- ↑ "Periventricular heterotopia | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Retrieved 2019-04-28.
- 1 2 "Arnold Chiari Malformation: Symptoms, Types, and Treatment". WebMD. Retrieved 2019-04-28.
- 1 2 Vissers, Lisenka E. L. M.; Gilissen, Christian; Veltman, Joris A. (2015-10-27). "Genetic studies in intellectual disability and related disorders". Nature Reviews Genetics. 17 (1): 9–18. doi:10.1038/nrg3999. ISSN 1471-0056. PMID 26503795. S2CID 16723395.