Glutathione synthetase deficiency
| Glutathione synthetase deficiency | |
|---|---|
| Other names | Pyroglutamicaciduria, 5-Oxoprolinuria, Oxoprolinase deficiency, Pyroglutamic aciduria |
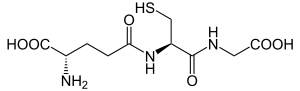 | |
| Glutathione | |
Glutathione synthetase deficiency (GSD) is a rare autosomal recessive[1] metabolic disorder that prevents the production of glutathione. Glutathione helps prevent damage to cells by neutralizing harmful molecules generated during energy production. Glutathione also plays a role in processing medications and cancer-causing compounds (carcinogens), and building DNA, proteins, and other important cellular components.
Genetics

Mutations in the GSS gene cause glutathione synthetase deficiency. This gene provides instructions for making the enzyme glutathione synthetase. This enzyme is involved in a process called the gamma-glutamyl cycle, which takes place in most of the body's cells. This cycle is necessary for producing a molecule called glutathione. Glutathione protects cells from damage caused by unstable oxygen-containing molecules, which are byproducts of energy production. Glutathione is called an antioxidant because of its role in protecting cells from the damaging effects of these unstable molecules which are byproducts of energy production. Mutations in the GSS gene prevent cells from making adequate levels of glutathione, leading to the signs and symptoms of glutathione synthetase deficiency.
This disorder is inherited in an autosomal recessive pattern, which means the defective gene is located on an autosome, and two copies of the gene - one from each parent - are required to be born with the disorder. The parents of an individual with an autosomal recessive disorder each carry one copy of the defective gene, but usually are not affected by the disorder.
Diagnosis
Glutathione synthetase deficiency can be classified into three types: mild, moderate and severe.[2]
- Mild glutathione synthetase deficiency usually results in the destruction of red blood cells (hemolytic anemia). Rarely, affected people also excrete large amounts of a compound called 5-oxoproline (also called pyroglutamic acid, or pyroglutamate) in their urine (5-oxoprolinuria). This compound builds up when glutathione is not processed correctly in cells.[2]
- Individuals with moderate glutathione synthetase deficiency may experience symptoms beginning shortly after birth including hemolytic anemia, 5-oxoprolinuria, and elevated acidity in the blood and tissues (metabolic acidosis).
- In addition to the features present in moderate glutathione synthetase deficiency, individuals affected by the severe form of this disorder may experience neurological symptoms. These problems may include seizures; a generalized slowing down of physical reactions, movements, and speech (psychomotor retardation); intellectual disability; and a loss of coordination (ataxia). Some people with severe glutathione synthetase deficiency also develop recurrent bacterial infections.
Treatment
As of 2018, there is no cure for GSD, and treatment is restricted to manage symptoms and associated problems.[3] Thus, sodium bicarbonate is recommended to treat metabolic acidosis, and antioxidants, among them vitamins E and C, can reduce oxidative damage.[3]
References
- ↑ Njålsson, Runa; Ristoff, Ellinor; Carlsson, Katarina; Winkler, Andreas; Larsson, Agne; Norgren, Svante (2005). "Genotype, enzyme activity, glutathione level, and clinical phenotype in patients with glutathione synthetase deficiency". Human Genetics. 116 (5): 384–9. doi:10.1007/s00439-005-1255-6. PMID 15717202. S2CID 19707969.
- 1 2 Njålsson, R. (2005). "Glutathione synthetase deficiency". Cellular and Molecular Life Sciences. 62 (17): 1938–45. doi:10.1007/s00018-005-5163-7. PMID 15990954. S2CID 59244.
- 1 2 NIH (May 13, 2018). "Glutathione synthetase deficiency". NIH/GARD. Retrieved January 24, 2020.
Further reading
- Glutathione synthetase deficiency at NLM Genetics Home Reference
- Beutler, E; Gelbart, T; Pegelow, C (1986). "Erythrocyte glutathione synthetase deficiency leads not only to glutathione but also to glutathione-S-transferase deficiency". Journal of Clinical Investigation. 77 (1): 38–41. doi:10.1172/JCI112298. PMC 423305. PMID 3944259.