Simpson–Golabi–Behmel syndrome
| Simpson–Golabi–Behmel syndrome | |
|---|---|
| Other names: Sara Agers syndrome, Golabi–Rosen syndrome, Simpson dysmorphia syndrome (SDYS) or X-linked dysplasia gigantism syndrome (DGSX)[1] | |
 | |
| Simpson–Golabi–Behmel syndrome has an X-linked recessive pattern of inheritance. | |
| Causes | Genetic |
Simpson–Golabi–Behmel syndrome (SGBS), is a rare inherited congenital disorder that can cause craniofacial, skeletal, cardiac, and renal abnormalities. The syndrome is inherited in an X-linked recessive fashion,[2] where males express the phenotype and females usually do not. Females that possess one copy of the mutation are considered to be carriers of the syndrome and may express varying degrees of the phenotype.
Types
There are two types of SGBS, each found on a different gene:
| Type | OMIM | Gene | Locus |
|---|---|---|---|
| SGBS1 | 312870 | GPC3 | Xq26 |
| SGBS2 | 300209 | OFD1 | Xp22 |
SGBS is also considered to be an overgrowth syndrome (OGS). OGS is characterized by a 2-3 standard deviation increase in weight, height, or head circumference above the average for sex and age. One of the most noted features of OGS is the increased risk of neoplasms in certain OGSs. SGBS in particular has been found to have a 10% tumor predisposition frequency with 94% of cases occurring in the abdominal region, most being malignant. It is common for tumors to be embryonal in type and appear before the age of 10. There are five different types of tumors that patients with SGBS might develop, all intra-abdominal: Wilms tumor, Hepatoblastoma, Hepatocarcinoma, Gonadoblastoma, and Neuroblastoma. The most common types of tumors developed in patients are the Wilms tumor and hepatoblastoma.[3]
Symptoms and signs
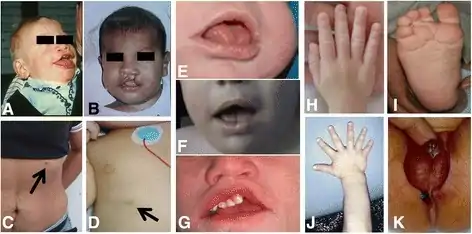
- Macrosomia
- Macroglossia
- Advanced bone age
- Organomegaly
- Neonatal hypoglycemia
- Neoplasms
- Congenital diaphragmatic hernia
- (protruding jaw and tongue, widened nasal bridge, upturned nasal tip)
- Hands and feet are short and broad with dysplastic nails
- Cutaneous syndactyly
- Polydactyly
- Pectus excavatum
- Talipes
- Vertebral segmentation defects
- Supernumerary nipples
- Structural and conductive cardiac defects
- Multicystic dysplastic kidneys
- Hypotonia
- Seizures
- Brain malformations
- Developmental disabilities
- Mental retardation- can be inexistent or mild to severe
Causes
Although not all causes of SGBS have been identified, one cause of SGBS type I is a mutation of the glypican-3 gene (GPC3) on the X chromosome locus q26.1. This particular gene is widely expressed, especially in tissues derived from the mesoderm during fetal development. The function of this gene is to produce a protein that acts as a cell surface receptor that binds to transcription factors. Binding of the transcription factors allows regulation of cellular responses to growth factors such as members of the hedgehog protein family. When large or small deletions and missense mutations occur along the GPC3 gene, GPC3 can no longer negatively regulate Hedgehog signaling during development, therefore increasing cell proliferation and the risk of developing cancer.[4] Limb patterning and skeletal development may also go awry when GCP3 mutations inhibit regulations of responses to bone morphogenetic proteins, another type of growth factor.[5]
It has been suggested that SGBS type II may be caused by duplication of the GPC4 gene, which helps to regulate cell division and growth.[6]
Also, some patients diagnosed with SGBS do not have any GPC3 or GPC4 deletions or mutations. Possible explanations include promoter mutation or silencing of the GPC3 gene causing reduced expression in these patients.[7]
Genetics
The disorder is passed on in an X-linked recessive fashion.
Diagnosis
Detection usually begins with a routine doctor visit when the fundal height is being measured or during an ultrasound examination. When large for gestational age fetuses (LGA) are identified, there are two common causes: maternal diabetes or incorrect dates. However, if these two causes can be ruled out, an ultrasound is performed to detect for overgrowth and other abnormalities. At this point, it becomes essential for a clinical geneticist to assist in the correct selection of tests and possible diagnosis.[8]
First signs of SGBS may be observed as early as 16 weeks of gestation. Aids to diagnosing might include the presence of macrosomia, polyhydramnios, elevated maternal serum-α-fetoprotein, cystic hygroma, hydrops fetalis, increased nuchal translucency, craniofacial abnormalities, visceromegaly, renal abnormalities, congenital diaphragmatic hernia, polydactyly, and a single umbilical artery.[6]
If there is a known mutation in the family, prenatal testing is available. Prenatal testing is also possible by looking for evidence of the mild SGBS phenotype in the mother and the positive SGBS phenotype in male family members. Family members who are positive of SGBS may undergo mutational analysis of genes GCP3, GCP4, and CXORF5. Genomic balance in Xp22 and Xq26 may also be analyzed through array comparative genomic hybridization.[6]
Due to the high percentage of male deaths during the neonatal period, early detection of tumors is crucial. In order to detect the presence of tumors, screening in SGBS patients should include abdominal ultrasound, urinalysis, and biochemical markers that screen for embryonic tumors.[8]
Once the infant is born, possibility of hypoglycemia must be assessed along with cardiac, genitalia, liver, and adrenal evaluations. Such tests include chest radiographs, electrocardiogram, echocardiogram, renal sonography, and abdominal sonography to test for possible abnormalities.[9]
Treatment
Since the syndrome is caused by a genetic mutation in the individual's DNA, a cure is not available. Treatment of the symptoms and management of the syndrome, however, is possible.
Depending on the manifestation, surgery, increased intake of glucose, special education, occupational therapy, speech therapy, and physical therapy are some methods of managing the syndrome and associated symptoms.[10]
Research
SGBS is similar to another overgrowth syndrome called Beckwith–Wiedemann syndrome.
SGBS Cells are a unique tool to study the function of Human adipocyte biology. These cells are similar to human primary preadipocytes, and may or may not become a popular model instead of Mouse 3T3-L1 cells to study the secretion and adipokine profile in the future. This cellular tool has been described and developed by Dr. Martin Wabitsch, University of Ulm, Germany.[11]
References
- ↑ Online Mendelian Inheritance in Man (OMIM): 312870
- ↑ Garganta CL, Bodurtha JN (1992). "Report of another family with Simpson–Golabi–Behmel syndrome and a review of the literature". Am J Med Genet. 44 (2): 129–135. doi:10.1002/ajmg.1320440202. PMID 1456279.
- ↑ Lapunzina, Pablo (15 August 2005). "Risk of tumorigenesis in overgrowth syndromes: A comprehensive review". American Journal of Medical Genetics Part C. 137C (1): 53–71. doi:10.1002/ajmg.c.30064. PMID 16010678. S2CID 24798488.
- ↑ Slavotinek, Anne M. (15 May 2007). "Single gene disorders associated with congenital diaphragmatic hernia". American Journal of Medical Genetics Part C. 145C (2): 172–183. doi:10.1002/ajmg.c.30125. PMID 17436300. S2CID 20749769.
- ↑ Debaun, Michael R.; Ess, Jennifer; Saunders, Scott (2001). "Simpson Golabi Behmel Syndrome: Progress toward Understanding the Molecular Basis for Overgrowth, Malformation, and Cancer Predisposition". Molecular Genetics and Metabolism. 72 (4): 279–86. doi:10.1006/mgme.2001.3150. PMID 11286501.
- 1 2 3 Chen, Chih-Ping (1 June 2012). "Prenatal findings and the genetic diagnosis of fetal overgrowth disorders: Simpson-Golabi-Behmel syndrome, Sotos syndrome, and Beckwith-Wiedemann syndrome". Taiwanese Journal of Obstetrics and Gynecology. 51 (2): 186–191. doi:10.1016/j.tjog.2012.04.004. PMID 22795092.
- ↑ Veugelers, M.4; Cat, BD; Muyldermans, SY; Reekmans, G; Delande, N; Frints, S; Legius, E; Fryns, JP; Schrander-Stumpel, C (22 May 2000). "Mutational analysis of the GPC3/GPC4 glypican gene cluster on Xq26 in patients with Simpson-Golabi-Behmel syndrome: identification of loss-of-function mutations in the GPC3 gene". Human Molecular Genetics. 9 (9): 1321–1328. doi:10.1093/hmg/9.9.1321. PMID 10814714.
- 1 2 Vora, Neeta; Bianchi, Diana W. (1 October 2009). "Genetic considerations in the prenatal diagnosis of overgrowth syndromes". Prenatal Diagnosis. 29 (10): 923–929. doi:10.1002/pd.2319. PMC 4426974. PMID 19609940.
- ↑ DeBaun, Michael R.; Ess, Jennifer; Saunders, Scott (1 April 2001). "Simpson Golabi Behmel Syndrome: Progress toward Understanding the Molecular Basis for Overgrowth, Malformation, and Cancer Predisposition". Molecular Genetics and Metabolism. 72 (4): 279–286. doi:10.1006/mgme.2001.3150. PMID 11286501.
- ↑ Golabi, Mahin (1993). "Simpson-Golabi-Behmel Syndrome Type 1". U.S. National Library of Medicine. Archived from the original on 2020-05-09. Retrieved 2020-11-06.
- ↑ Wabitsch, Martin (January 2001). "Characterization of a human preadipocyte cell strain with high capacity for adipose differentiation". International Journal of Obesity and Related Metabolic Disorders. 25 (1): 8–15. doi:10.1038/sj.ijo.0801520. PMID 11244452.
External links
- Simpson–Golabi–Behmel syndrome at NIH's Office of Rare Diseases
- GeneReview/NCBI/NIH/UW entry on Simpson–Golabi–Behmel Syndrome Archived 2010-06-01 at the Wayback Machine
| Classification | |
|---|---|
| External resources |
|