مرض هارتناب
مرض هارتناب (بالإنجليزية: Hartnup disease) (المعروف أيضاً باسم «الالتهاب الجلدي الشبيه بالبلاغرا»[1] أو«اضطراب هارتناب»[2]) هو مرض وراثي مُتنحٍّ[3] يتمثل باضطراب أيضيّ يؤثر على امتصاص الأحماض الأمينية غير القطبية (خصوصاً التريبتوفان الذي من الممكن أن يتحول إلى سيروتونين، وَميلاتونين، وَنياسين)، وَيُعتبر النياسين هو العنصر الأولي لـنيكوتيناميد، الذي يعتبر بدوره مكوّن ضروري من مكونات ثنائي نوكليوتيد الأدنين وأميد النيكوتين NAD+[4]:541.
| مرض هارتناب | |
|---|---|
| معلومات عامة | |
| الاختصاص | علم الغدد الصم |
| من أنواع | اضطراب التمثيل الغذائي للحمض الأميني ، وجلاد ضوئي، ومرض |
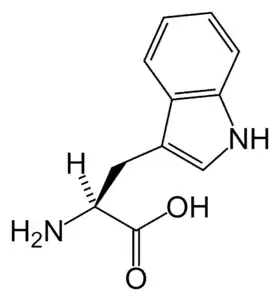

يقع الجين المسبب للمرض -المسمى SLC6A19- على الكروموسوم الخامس.[5].وسمي على اسم العائلة الإنجليزية، هارتناب، التي عانت من هذا المرض.
الأعراض والعلامات
تظهر علامات مرض هارتناب أثناء مرحلة الطفولة بشكل متباين، ومن هذه الأعراض: فشل الطفل في النمو كأقرانه، وحدوث حساسية عالية للضوء، ونوبات من الرنح أو عدم القدرة على التحكم في تنسيق حركات العضلات الإرادية والرجفان.
يُعتبر النيكوتين اميد من المكونات الضرورية لإنتاج ناقلات الأحماض الأمينية المحايدة في الأنابيب الكلوية الدانية في الكلى وكذلك في خلايا الغشاء المخاطي في الأمعاء الدقيقة، ولذلك فإن الأعراض المصاحبة لهذا الاضطراب تنتج عن فقدان كميّات كبيرة من الأحماض الأمينية في البول.
يُعتبر مرض البلاغرا أو الحُصاف الجلديّ مرض مُشابه لهذا الاضطراب حيثُ ينتج عنه التهاب في الجلد وحالات من الخرف أو فقدان الذاكرة والإسهال وينتج الحُصاف أيضاً بسبب انخفاض نسبة النيكوتين اميد.
ينتج مرض هارتناب بشكل أساسي بسبب مشاكل في نقل الأحماض الأمينية وتحركاتها في الأمعاء والكليتين وإلا فإن كليهما سيعمل بشكل طبيعي، أما آثار المرض فأكثر ما تظهر على الدماغ وَالجلد، حيثُ تبدأ الأعراض بالظهور في فترة الطفولة المبكرة وقد تستمر في الظهور لمرحلة البلوغ، وَيُحفز ظهورَ هذه الأعراض الكثير من العوامل؛ أَهمها ضوء الشمس والحرارة والضغط النفسي واستعمال بعض الأدوية، أما بالنسبة للغذاء فإن مرحلة سوء التغذية دائماً تسبق حدوث المرض، وتبدأ نوبات المرض بالانحدار مع العمر وتحدث النوبات عادةً بشكل مُتقطع بسبب نقص النيكوتين اميد (النياسين اميد).
عادةً ما يظهر الطفح الجلدي على أجزاء الجسم المكشوفة للشمس، ومن المشاكل التي تُصاحب مرض هارتناب: التخلف العقلي وقصر القامة والصداع المستمر ونوبات من الانهيار وبعض المشاكل النفسية مثل الهلوسات وتقلب المزاج.[6]
الأسباب
يعد مرض هارتنب من الأمراض الوراثية المتنحية فهو لا يحدث إلا عند اجتماع الجينات أثناء التلقيح من كلا الأبوين ومن أحد أسبابه زواج الأقارب.
تم اكتشاف اختلالات في نقل بعض الأحماض الأمينية سجل ذلك عام 1960 بسبب ارتفاع الأيض البكتيري لحمض التريبتوفان (الاندول)والتريبتوفان في البول عند المرضى كجزء من فقد الأحماض الأمينية في البول بشكل عام في هذا المرض. وقد كانت خسارة كميات كبيرة من التربتوفان -بسبب سوء الامتصاص- السبب في الأعراض المشابهة بين مرض هارتناب ومرض البلاغرا.و يبدو من الدراسات التي تتناول في موضوعها التربتوفان أنه كان هناك مشكلة عامة في نقل الأحماض الأمينية.[7]
في عام 2004، وضع جين مسبب للمرض وهو جين SLC6A195 في نطاق جين 5P15.33. وهذا الجين هو أحد الجينات المسؤولة عن نقل الأحماض الأمينية المتعادلة ويعتمد في عمله على الصوديوم بالتحديد ويوجد هذا الناقل في الكليتين والغشاء المبطن للأمعاء الدقيقة.[8]
التشخيص
إن الجين المسبب للمرض في طبيعته يتحكم بامتصاص بعض الأحماض الأمينية من الأمعاء وبإعادة امتصاصها من الكلى، وبناء على ذلك فإن الأشخاص المصابين غير قادرين على امتصاصها من الأعماء وإعادة امتصاص تلك الأحماض من الكلى؛ مما يجعلها تخرج في البول ليظل الجسم يعاني من نقص في الحموض الأمينية والتي تعد الوحدات الأساسية لبناء البروتينات. كما وإن كمية التربتوفان القليلة في الدم تجعل الجسم غير قادرعلى تصنيع كميات كافية من مركبات فيتامين ب، خاصة عند الحاجة الشديدة إلى مزيد من الفيتامينات[6]
العلاج
يمكن التغلب على مشكلة قلة الحموض الأمنية من خلال نظام غذائي يحتوي على كميات كبيرة من البروتين، أما قلة التغذية فإنها تزيد من حدة نوبات المرض وأعراضه. ينصح الأطباءُ الأشخاص الذين يعانون من أعراض المرض باستخدام مستحضرات ومواد كيميائية أو فيزيائية للوقاية من ضوء الشمس كاستعمال واقيات الشمس بدرجة Spf) 15) أو أكثر. كما وعلى المصابين تجنب أية أدوية تزيد من حساية الجسم للضوء. وللأشخاص الذين يعانون من نقص في النيكوتين اميد فإنه يتم تزويدهم بكميات يومية منه للتقليل من أعراض ونوبات المرض. وأما الأشخاص الذين يعانون من مشاكل عصبية أو نفسية بسبب المرض فيجب أن يتم علاجهم أيضا.[8]
انظر أيضاً
مراجع
- Rapini, Ronald P.؛ Bolognia, Jean L.؛ Jorizzo, Joseph L. (2007)، Dermatology: 2-Volume Set، St. Louis: Mosby، ISBN 1-4160-2999-0.
- الوراثة المندلية البشرية عبر الإنترنت (OMIM): 234500
- Kleta R, Romeo E, Ristic Z, Ohura T, Stuart C, Arcos-Burgos M, Dave MH, Wagner CA, Camargo SR, Inoue S, Matsuura N, Helip-Wooley A, Bockenhauer D, Warth R, Bernardini I, Visser G, Eggermann T, Lee P, Chairoungdua A, Jutabha P, Babu E, Nilwarangkoon S, Anzai N, Kanai Y, Verrey F, Gahl WA, Koizumi A (سبتمبر 2004)، "Mutations in SLC6A19, encoding B0AT1, cause Hartnup disorder"، Nature Genetics، 36 (9): 999–1002، doi:10.1038/ng1405، PMID 15286787.
- James, William D.؛ Berger, Timothy G.؛ وآخرون (2006)، Andrews' Diseases of the Skin: clinical Dermatology، Saunders Elsevier، ISBN 0-7216-2921-0.
- Seow HF, Brer S, Brer A, Bailey CG, Potter SJ, Cavanaugh JA, Rasko JE (سبتمبر 2004)، "Hartnup disorder is caused by mutations in the gene encoding the neutral amino acid transporter SLC6A19"، Nature Genetics، 36 (9): 1003–7، doi:10.1038/ng1406، PMID 15286788.
- "Hartnup Disease"، مؤرشف من الأصل في 5 نوفمبر 2010، اطلع عليه بتاريخ 23 نوفمبر 2008.
- Milne, M.D., Crawford, M.A., Girao, C.B. and Loughridge, L. (1961) The metabolic disorder of the Hartnup disease. Q. J. Med. 29: 407-421
- Lidija Kandolf Sekulovic، "Hartnup Disease"، مؤرشف من الأصل في 23 نوفمبر 2008، اطلع عليه بتاريخ 23 نوفمبر 2008.