Atrioventricular septal defect
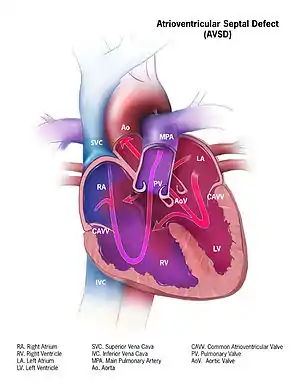
Atrioventricular septal defect (AVSD) or atrioventricular canal defect (AVCD), also known as "common atrioventricular canal" or "endocardial cushion defect" (ECD), is characterized by a deficiency of the atrioventricular septum of the heart that creates connections between all four of its chambers. It is a very specific combination of 3 defects:
| Atrioventricular septal defect | |
|---|---|
| Other names | Atrioventricular canal defect (AVCD), endocardial cushion defect (ECD) |
 | |
| Specialty | Cardiology |
| Symptoms | Heart failure; pulmonary hypertension; dyspnea; cyanosis; mitral regurgitation |
| Types | Partial, Incomplete, Complete, Transitional |
| Risk factors | Family history of congenital heart disease |
| Diagnostic method | Ultrasound and echocardiography |
| Treatment | Open heart surgery |
1) Atrial Septal Defect (ASD), a hole in the wall between the right and left atria;
2) Ventricular Septal Defect (VSD), a hole in the wall between the right and left ventricles; and
3) Abnormalities of the mitral and/or tricuspid valves.[1][2]
AVCD is caused by an abnormal or inadequate fusion of the superior and inferior endocardial cushions with the mid portion of the atrial septum and the muscular portion of the ventricular septum.[3] Unlike some heart defects, the condition will not resolve over time and most infants must undergo open heart surgery. The surgery to correct this defect is usually successful and most babies do very well post-op.[4]
Symptoms and signs
Symptoms may include difficulty breathing (dyspnea) and bluish discoloration on skin, fingernails, and lips (cyanosis).[5] An infant will begin to show signs of congestive heart failure, which can include rapid breathing, feeding problems, slow weight gain, low energy, and cold, clammy sweating.[4] Symptoms often appear between 1-2 months of age but can occur earlier in some newborns.[4]
Complications
Normally, the four chambers of the heart divide oxygenated and de-oxygenated blood into separate pools. When holes form between the chambers, as in AVSD, the pools can mix. Consequently, arterial blood supplies become less oxygenated than normal, causing ischemia and cyanosis in distal tissues.[3] To compensate, the heart must pump a larger volume of blood to deliver enough oxygen, leading to cardiac enlargement and hypertrophy.[5]
The development of pulmonary hypertension is very serious. And this because the left ventricle is weakened due to its overuse. When this happens, the pressure backs up into the pulmonary veins and the lungs.[5] This type of damage is irreversible which is why immediate treatment is recommended after diagnosis.[6]
Associated conditions
Down syndrome is often associated with AVCD.[7] Other risk factors include: having a parent with a congenital heart defect, alcohol use while pregnant, uncontrolled diabetes treatment during pregnancy and some medications during pregnancy.[5]
This type of congenital heart defect is associated with patients with Down syndrome (trisomy 21) or heterotaxy syndromes.[8] 45% of children with Down syndrome have congenital heart disease. Of these, 35–40% have AV septal defects.[9] Approximately 40-50% of fetuses diagnosed with AVCD have Down syndrome, and a further 15-20% are associated with other chromosomal abnormalities and syndromes, such as DiGeorge syndrome.[3][10] The remaining 30-40% of cases are not linked to a syndrome, with AVCD observed without other major defects.
AVCD is also linked with Noonan syndrome.[3] The pattern seen in those patients with Noonan syndrome differ from those patients who have Down syndrome in that "partial" AVCD is more prevalent in those with NS, whereas those with down syndrome show a prevalence of the "complete" form of AVCD.[11]
Pathophysiology

Defective embryonic formation of the heart results in multiple holes between the heart chambers. In AVSD, all four chambers are connected, but the exact characteristics of holes and malformations may vary between patients. Even within the categories of "complete" and "partial" AVSD, multiple morphologies exist, with varying clinical consequences. Clinical and physiological manifestations of disease may also change over time, in response to continued stress.[3]
Genetic Relationship
Like other congenital heart defects, major associations have been found between AVCD and genes regulating embryonic cell cilia.[10] These human cell cilia normally contain receptors for signal molecules that regulate the healthy and organized tissue. Dysfunctional cilia can create multiple disease manifestations, leading to broad syndromes.[10] Chromosome 21 harbors important regulators for cilia, and trisomy 21 (Down syndrome) can de-regulate them.[12]
Diagnosis
AVSDs can be detected by cardiac auscultation; they cause atypical murmurs and loud heart tones. Confirmation of findings from cardiac auscultation can be obtained with a cardiac ultrasound (echocardiography - less invasive) and cardiac catheterization (more invasive). It is also possible to diagnose AVSD in-utero via routine fetal ultrasounds or, more conclusively, fetal echocardiograms.[3]
Classification
A variety of different classifications have been used, but the defects are usefully divided into "partial" and "complete" forms.
- In the partial AVSD, there is a small or partial defect in the interventricular septum, and a primum atrial septal defect, which is a moderate or large connection between the atria, often featuring mitral valve regurgitation.[3] Partial AVSD may be asymptomatic in early childhood, but typically progresses by late childhood or adulthood into symptoms of heart failure. The onset of symptoms may be earlier in children with more significant mitral regurgitation.[3]
- In the complete AVSD (CAVSD), there is a large ventricular component beneath either or both the superior or inferior bridging leaflets of the AV valve. The defect involves the whole area of the junction of the upper and lower chambers of the heart, i.e. where the atria join the ventricles. There is a large hole between the lower portion of the atria and the upper or 'inlet' portion of the ventricles and this is associated with a significant abnormality of the valves separating the atria from the ventricles. The valves in effect become a common atrio-ventricular valve, and the severity of the defect depends largely on the supporting attachments of the valve to the ventricles and whether the valve allows dominant flow from the right atrium to right ventricle and from left atrium to left ventricle ("unbalanced" flow). The overall problems are similar to those of VSD but are more complicated. There is an increased flow of blood to the lungs through both the ventricular and atrial components of the defect. In addition, the abnormal atrio-ventricular valve invariably leaks, so that when the ventricles contract, blood flows not only forwards to the body and the lungs, but also backwards into the atria. The back-pressure effect on the atria causes congestion of blood in the left atrium in particular, and this in turn causes congestion in the veins draining the lungs. The effect on the baby is to worsen the heart failure that is associated with an isolated VSD and to hasten the onset of pulmonary hypertension. It should be mentioned that CAVSD is found in approximately one-third of babies who have Down syndrome, but it also occurs as an isolated abnormality.
Treatment
Treatment is surgical and involves closure of the atrial and ventricular septal defects and restoration of a competent left AV valve as far as is possible. Open surgical procedures require a heart-lung machine and are done with a median sternotomy. Surgical mortality for uncomplicated ostium primum defects in experienced centers is 2%; for uncomplicated cases of complete atrioventricular canal defect, 4% or less. Certain complications such as tetralogy of Fallot or highly unbalanced flow across the common AV valve can increase risk significantly.[13][14]
Infants born with AVSD are generally in sufficient health to not require immediate corrective surgery. If surgery is not required immediately after birth, the newborn will be closely monitored for the next several months, and the operation held-off until the first signs of lung distress or heart failure. This gives the infant time to grow, increasing the size of, and thereby the ease of operation on, the heart, as well as the ease of recovery. Infants will generally require surgery within three to six months, however, they may be able to go up to two years before the operation becomes necessary, depending on the severity of the defect.[15]
See also
References
- "Atrioventricular Septal Defect - Seattle Children's". Seattle Children’s Hospital. Retrieved 2023-03-27.
- "Atrioventricular Septal Defect (AVSD)". Children's Minnesota. Retrieved 2023-03-27.
- Fleishman CE, Tugertimur A (December 2022). Triedman JK, Armsby C (eds.). "Clinical manifestations and diagnosis of atrioventricular (AV) canal defects". UpToDate. Retrieved 2023-01-05.
- "Atrioventricular Septal Defect or AV Canal (AVSD)". C.S. Mott Children’s Hospital, University of Michigan Health. Retrieved 24 March 2023.
- "Atrioventricular canal defect - Symptoms and causes". Mayo Clinic. 2022-11-29. Archived from the original on 29 November 2022. Retrieved 2023-01-05.
- Calabrò R, Limongelli G (April 2006). "Complete atrioventricular canal". Orphanet Journal of Rare Diseases. 1: 8. doi:10.1186/1750-1172-1-8. PMC 1459121. PMID 16722604.
- Irving CA, Chaudhari MP (April 2012). "Cardiovascular abnormalities in Down's syndrome: spectrum, management and survival over 22 years". Archives of Disease in Childhood. 97 (4): 326–330. doi:10.1136/adc.2010.210534. PMID 21835834.
- Fyler DC, Buckley LP, Hellenbrand WE, Cohn HE, Kirklin JW, Nadas AS, Cartier JM, Breibart MH (1980). "Report of the New England Regional Infant Cardiac Program". Pediatrics. 65 (suppl): 441–444.
- Al-Hay AA, MacNeill SJ, Yacoub M, Shore DF, Shinebourne EA (February 2003). "Complete atrioventricular septal defect, Down syndrome, and surgical outcome: risk factors". The Annals of Thoracic Surgery. 75 (2): 412–421. doi:10.1016/s0003-4975(02)04026-2. PMID 12607648.
- Pugnaloni F, Digilio MC, Putotto C, De Luca E, Marino B, Versacci P (May 2020). "Genetics of atrioventricular canal defects". Italian Journal of Pediatrics. 46 (1): 61. doi:10.1186/s13052-020-00825-4. PMC 7222302. PMID 32404184.
- Marino B, Digilio MC, Toscano A, Giannotti A, Dallapiccola B (December 1999). "Congenital heart diseases in children with Noonan syndrome: An expanded cardiac spectrum with high prevalence of atrioventricular canal". The Journal of Pediatrics. 135 (6): 703–706. doi:10.1016/S0022-3476(99)70088-0. PMID 10586172.
- Galati DF, Sullivan KD, Pham AT, Espinosa JM, Pearson CG (September 2018). "Trisomy 21 Represses Cilia Formation and Function". Developmental Cell. 46 (5): 641–650.e6. doi:10.1016/j.devcel.2018.07.008. PMC 6557141. PMID 30100262.
- Kirklin J, Barratt-Boyes B, eds. (1986). Cardiac Surgery. New York: Wiley. pp. 463–497.
- Marx GR, Fyler DC (2006). "Endiocardial Cusion Defects". In Keane JF, Lock JE, Fyler DC (eds.). Pediatric Cardiology (2nd ed.). Philadelphia: Saunders-Elsevier. pp. 663–674. ISBN 978-1-4160-2390-6.
- Hay WW, Levin MJ, Sondheimer JM, Deterding RR (2007). Current pediatric diagnosis & treatment (18th ed.). New York: Lange Medical Books/McGraw-Hill, Medical Pub. Division. ISBN 978-0-07-146300-3.