Brugada syndrome
Brugada syndrome (BrS) is a genetic disorder in which the electrical activity of the heart is abnormal due to channelopathy.[2] It increases the risk of abnormal heart rhythms and sudden cardiac death.[2] Those affected may have episodes of syncope.[2] The abnormal heart rhythms seen in those with Brugada syndrome often occur at rest.[1][5] They may be triggered by a fever.[1][5]
| Brugada syndrome | |
|---|---|
| Other names | Sudden unexplained nocturnal death syndrome, bangungut, pokkuri death syndrome[1] |
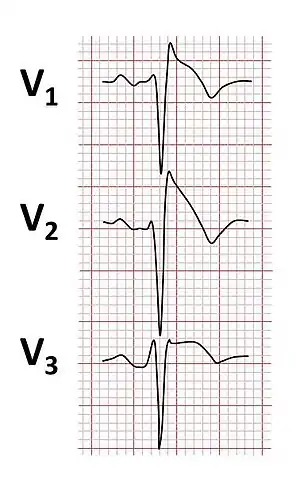 | |
| Typical type 1 ECG changes seen in Brugada syndrome | |
| Specialty | Cardiology |
| Symptoms | Passing out, sudden cardiac death[2] |
| Usual onset | Adulthood[2] |
| Causes | Genetics, certain medication[2] |
| Risk factors | Family history, Asian descent, male[1][2] |
| Diagnostic method | Electrocardiogram (ECG), genetic testing[2][3] |
| Differential diagnosis | Romano-Ward syndrome, arrhythmogenic cardiomyopathy, Duchenne muscular dystrophy[3] |
| Treatment | Watchful waiting, implantable cardioverter defibrillator (ICD)[3][4] |
| Frequency | 1 per 2000[1] |
| Deaths | 8% of sudden cardiac death[2] |
About a quarter of those with Brugada syndrome have a family member who also has the condition.[2] Some cases may be due to a new genetic mutation or certain medications.[1] The most commonly involved gene is SCN5A which encodes the cardiac sodium channel.[6] Diagnosis is typically by electrocardiogram (ECG), however, the abnormalities may not be consistently present.[2] Medications such as ajmaline may be used to reveal the ECG changes.[2] Similar ECG patterns may be seen in certain electrolyte disturbances[7] or when the blood supply to the heart has been reduced.[8]
There is no cure for Brugada syndrome.[3] Those at higher risk of sudden cardiac death may be treated using an implantable cardioverter defibrillator (ICD).[4] In those without symptoms the risk of death is much lower, and how to treat this group is less clear.[3][9] Isoproterenol may be used in the short term for those who have frequent life-threatening abnormal heart rhythms, while quinidine may be used longer term.[3][10] Testing people's family members may be recommended.[3]
The condition affects between 1 and 30 per 10,000 people.[2] It is more common in males than females and in those of Asian descent.[1][2] The onset of symptoms is usually in adulthood.[2] It was firstly described by Andrea Nava and Bortolo Martini in Padova in 1989 [48] but it is named after the Catalan cardiologists Pedro and Josep Brugada who described the condition in 1992.[3][11] Chen first described the genetic abnormality of SCN5A channels [49]
Signs and symptoms
While many of those with Brugada syndrome do not have any symptoms, Brugada syndrome may cause fainting or sudden cardiac death due to serious abnormal heart rhythms such as ventricular fibrillation or polymorphic ventricular tachycardia.[9] Blackouts may be caused by brief abnormal heart rhythms that revert to a normal rhythm spontaneously. If a dangerous heart rhythm does not stop by itself and is left untreated, the person may have a fatal cardiac arrest. However, blackouts can occur in those with Brugada syndrome despite a normal heart rhythm due to a sudden drop in blood pressure, known as vasovagal syncope.[2]
The abnormal heart rhythms seen in Brugada syndrome often occur at rest, following a heavy meal, or even during sleep.[5] These situations are linked to periods when the vagus nerve is activated, referred to as periods of high vagal tone. Abnormal heart rhythms may also occur during fever or following excessive alcohol. Sodium channel blocking medications, commonly used to treat cardiac arrhythmia, may also worsen the tendency to abnormal heart rhythms in patients with Brugada syndrome and should be avoided.[12][13]
Causes
The individual heart muscle cells communicate with each other with electrical signals that are disrupted in those with Brugada syndrome. As a genetic condition, the syndrome is ultimately caused by changes to a person's DNA, known as genetic mutations. The first mutations described in association with Brugada syndrome were in a gene responsible for a protein or ion channel that controls the flow of sodium ions through the cell membrane of heart muscle cells – the cardiac sodium channel. Many of the genetic mutations that have subsequently been described in association with Brugada syndrome influence the sodium current in some way, or affect other ionic currents.[8]
A long list of factors that can generate a Brugada ECG pattern have been described, including certain medications, electrolyte disturbances such as a decrease in the levels of potassium in the blood, and a reduction in blood supply to key areas of the heart, specifically the right ventricular outflow tract.[8] Drugs that have been implicated include antiarrhythmic medications such as flecainide, verapamil and propranolol, antidepressants such as amitryptiline, and drugs that enhance vagal tone such as acetylcholine. The ECG pattern can also be seen following excessive use of alcohol or cocaine.[8]
Genetics
Brugada syndrome is inherited in an autosomal dominant manner, meaning that only one copy of the defective gene is needed to produce the syndrome. However, a person diagnosed with the condition may be the first in their family to have Brugada syndrome if it has arisen as a new mutation.[5] The gene in which mutations are most commonly found in Brugada syndrome, known as SCN5A, is responsible for the cardiac sodium channel. Mutations in SCN5A associated with Brugada syndrome generally cause the flow of sodium ions to decrease. However, only 20% of cases of Brugada syndrome are associated with mutations in SCN5A, as in the majority of patients with Brugada syndrome genetic testing is unable to identify the genetic mutation responsible.[5] Over 290 mutations in the SCN5A gene have been discovered to date, each altering sodium channel function in subtly different ways.[14] This variation partially explains the differences in severity of the condition between different persons, ranging from a highly dangerous condition causing death at a young age to a benign condition that may not cause any problems at all. However, the genetics of Brugada syndrome are complex, and it is likely that the condition results from the interactions of many genes. Because of these complex interactions, some members of a family who carry a particular mutation may show evidence of Brugada syndrome while other carrying the same mutation may not, referred to as variable penetrance.[15] Mutations in the SCN5A gene seem to have a prognostic value.[16][17][18]
Several other genes have been identified in association with Brugada syndrome. Some are responsible for other proteins that form part of the sodium channel, known as sodium channel β subunits (SCN1B, SCN2B, SCN3B) while others form different types of sodium channel (SCN10A). Some genes encode ion channels that carry calcium or potassium ions (CACNA1C, CACNB2, KCND3, KCNE3, KCNJ8, KCNT1),[19] while others generate proteins that interact with ion channels. (GPD1L, PKP2, MOG1, FGF12). Another gene associated with this condition is RRAD.[20] The genes associated with Brugada syndrome and their description include:
| Type | OMIM | Gene | Notes |
|---|---|---|---|
| BrS1 | 601144 | SCN5A | NaV1.5 – α subunit of the cardiac sodium channel carrying the sodium current INa.[6] |
| BrS2 | 611778 | GPD1L | Glycerol-3-phosphate dehydrogenase like peptide – reduced GPD1-L activity leads to phosphorylation of NaV1.5 and decreased INa.[6] |
| BrS3 | 114205 | CACNA1C | CaV1.2 – α subunit of voltage-dependent calcium channel carrying the L-type calcium current ICa(L).[21] |
| BrS4 | 600003 | CACNB2 | CaVβ2B – β-2 subunit of the voltage-gated calcium channel carrying the L-type calcium current ICa(L).[21] |
| BrS5 | 600235 | SCN1B | NaVβ1 – β-1 subunit of the sodium channel carrying the sodium current INa.[21] |
| BrS6 | 604433 | KCNE3 | MiRP2 – β subunit to voltage-gated potassium channels. Modulates the transient outward potassium current Ito.[21] |
| BrS7 | 608214 | SCN3B | NaVβ3 – β-3 subunit of the cardiac sodium channel carrying the sodium current INa.[6] |
| BrS8 | 600935 | KCNJ8 | Kir6.1, carries the inward rectifier potassium current IKir.[6] |
| BrS9 | 114204 | CACNA2D1 | α2δ subunit of the voltage-gated calcium channel carrying the L-type calcium current ICa(L).[6] |
| BrS10 | 605411 | KCND3 | KV4.3, α-subunit of the transient outward potassium channel Ito.[6] |
| BrS11 | 607954 | RANGRF | Encodes MOG1 – influences trafficking of NaV1.5.[6] |
| BrS12 | 602701 | SLMAP | Sarcolemmal membrane–associated protein, a component of T-tubules and the sarcoplasmic reticulum – influences trafficking of NaV1.5.[6] |
| BrS13 | 601439 | ABCC9 | SUR2A, the adenosine triphosphate (ATP)-binding cassette transporter of the IK(ATP) channel.[6] |
| BrS14 | 601327 | SCN2B | NaVβ2 – Beta-2 subunit of the cardiac sodium channel carrying the sodium current INa.[6] |
| BrS15 | 602861 | PKP2 | Plakophillin-2 – interacts with INa.[6] |
| BrS16 | 601513 | FGF12 | Fibroblast growth factor homologous factor-1 – mutation decreases INa.[6] |
| BrS17 | 604427 | SCN10A | NaV1.8 – α subunit of the neuronal sodium channel.[21][22][23][24] |
| BrS18 | 604674 | HEY2 | Transcription factor identified in genome-wide association study.[6] |
| BrS19 | 603961 | SEMA3A | Semaphorin.[6] |
| BrS20 | 601142 | KCNAB2 | KVβ2, voltage-gated potassium channel β2 subunit - mutation increases Ito.[21] |
Some mutations associated with Brugada syndrome can also cause other heart conditions. Those who show more than one cardiac conditions at the same time caused by a single mutation are described as having an 'overlap syndrome'. An example of an overlap syndrome is Brugada and long QT syndrome (LQT3) caused by a mutation in SCN5A that reduces the peak sodium current but simultaneously leaves a persistent current leak.[8] Brugada syndrome has been described as overlapping with arrhythmogenic right ventricular cardiomyopathy (ARVC) caused by a mutation in the PKP2 gene, causing a Brugada ECG pattern but structural changes in the heart characteristic of ARVC.[25] Another example of an overlap syndrome would be Brugada syndrome and major aortopulmonary collateral arteries (MAPCAs) caused by a mutation in KCNT1 that leads to an abnormal gain-of-function in potassium channels of neurons and cardiomyocytes.[26][27]
Mechanisms

The abnormal heart rhythms seen in those with Brugada syndrome are typically dangerous arrhythmias such as ventricular fibrillation or polymorphic ventricular tachycardia, but those with BrS are also more likely to experience rapid heart rates due to less dangerous arrhythmias such as AV nodal re-entrant tachycardia[28] and abnormally slow heart rhythms such as sinus node dysfunction.[29] There are several mechanisms by which the genetic mutations causing this condition might produce these arrhythmias.[30]
Some argue that the main reason these arrhythmias arise is due to abnormally slow electrical conduction in areas of the heart, specifically the right ventricle. The genetic variants associated with BrS support the concept as SCN5A, the gene most commonly associated with the condition, along with SCN10A, SCN1B, SCN2B and SCN3B, all directly affect the sodium current INa. The sodium current is a major contributor to the characteristic flow of electrical charge across the membrane of heart muscle cells that occurs with each heartbeat known as the action potential. INa causes the initial rapid upstroke of the action potential (phase 0), and decreasing the early peak current, as occurs in BrS-associated genetic variants, leads to slowing of the electrical conduction through the heart muscle.[8] This slow conduction allows 'short circuits' to form, blocking the waves of electrical activity in some areas while allowing the waves to pass in others in a phenomenon known as wavebreak. Given the right circumstances, this wavebreak can allow the waves of electricity to perform a U-turn within the muscle, travelling in the reverse direction before beginning to rapidly circle around a point, referred to as re-entry, and causing an abnormal heart rhythm.[8] Those who support this view (known as the depolarisation hypothesis) argue that conduction slowing may explain why arrhythmias in those with Brugada syndrome tend to occur in middle age, when other factors such as scarring or fibrosis that accompany old age have exacerbated the tendency to conduction slowing caused by the genetic mutation.[30]
Others suggest that the main cause of arrhythmias is a difference in the electrical properties between the inside (endocardium) and outside (epicardium) of the heart (known as the repolarisation hypothesis).[8] The shape of the action potential differs between the epicardium and the endocardium. The action potential in cells from the epicardium shows a prominent notch after the initial spike due to a transient outward current. This notch is far less evident in cells from the endocardium, and the difference between the endocardium and epicardium are most clearly seen in the right ventricle. In those with Brugada syndrome, these differences are increased, creating a brief period within each cardiac cycle when current flows from the endocardium to the epicardium creating the characteristic ECG pattern. The differences in electrical properties between the epi- and endocardium are described as a 'transmural dispersion of repolarisation" which if large enough can lead to electrical impulses becoming blocked in some regions but not others. Once again, this wavebreak can allow the waves of electricity which usually travel in only one direction to instead begin circling around a point as a re-entrant circuit, causing an arrhythmia.[8]
A further factor promoting arrhythmias in Brugada syndrome is changes to the structure of the heart.[30] Whilst the heart of those with Brugada syndrome may look normal, scarring or fibrosis is often seen in particular regions of the heart, specifically the right ventricular outflow tract. As Brugada syndrome can be caused by mutation in many different genes, it is possible that different mechanisms may be responsible for the arrhythmias seen in different patients.[30]
Diagnosis
Electrocardiography

Brugada syndrome is diagnosed by identifying characteristic patterns on an electrocardiogram.[12] The pattern seen on the ECG includes ST elevation in leads V1-V3 with a right bundle branch block (RBBB) appearance. There may be evidence of a slowing of electrical conduction within the heart, as shown by a prolonged PR interval. These patterns may be present all the time, but may appear only in response to particular drugs (see below), when the person has a fever, during exercise, or as a result of other triggers. The ECG pattern may become more obvious by performing an ECG in which some of the electrodes are placed in different positions from usual, specifically by placing leads V1 and V2 higher up the chest wall in the 1st or 2nd intercostal spaces.[31]
Three forms of the Brugada ECG pattern have historically been described,[32] although the Type 3 pattern is frequently merged with the Type 2 pattern in contemporary practice.[33]
- Type 1 has a coved type ST elevation with at least 2 mm (0.2 mV) J-point elevation and a gradually descending ST segment followed by a negative T-wave.[33]
- Type 2 has a saddle-back pattern with at least 2 mm J-point elevation and at least 0.5 mm elevation of the terminal ST segment with a positive or biphasic T-wave.[33] A type 2 pattern can occasionally be seen in healthy subjects.
- Type 3 has a saddle-back (type 2 like) pattern, with at least 2 mm J-point elevation but less than 1 mm elevation of the terminal ST segment.[32]
According to current recommendations, only a Type 1 ECG pattern, occurring either spontaneously or in response to medication, can be used to confirm the diagnosis of Brugada syndrome as Type 2 and 3 patterns are not infrequently seen in persons without the disease.[9]
Provocation testing
Some medications, particularly antiarrhythmic drugs that block the cardiac sodium current INa, can reveal a Type 1 Brugada pattern in susceptible people. These drugs can be used to help make a diagnosis in those suspected of having Brugada syndrome (e.g. survivors of an unexplained cardiac arrest, family members of a person with Brugada syndrome) but in whom a diagnostic ECG pattern has not been seen.[2] In these cases, sodium current blocking medications can be given in a controlled environment.[31] The most commonly used drugs for this purpose are ajmaline, flecainide, and procainamide, with some suggestions indicating that ajmaline may be the most effective.[34] Precaution must be taken in giving these medications as there is a small risk of causing abnormal heart rhythms.[31]
Genetic testing
Genetic testing can be helpful to identify patients with Brugada syndrome, most commonly in family members of a person with Brugada syndrome, but sometimes performed in a person who has died suddenly and unexpectedly.[5] However, interpretation of the results of genetic testing is challenging. In family members who all carry a particular genetic variant associated with Brugada syndrome, some family members may show evidence of Brugada syndrome on their ECGs while others may not.[5] This means that carrying a genetic mutation associated with Brugada syndrome does not necessarily imply that a person is truly affected by the condition. To further complicate matters, many frequently occurring variations in the SCN5A gene do not cause any problems, and therefore genetic variants are sometimes identified in persons with Brugada syndrome that are not truly causing the disease.[35]
Other investigations
Invasive electrophysiological studies, in which wires are passed through a vein to stimulate and record electrical signals from the heart, can sometimes be used to assess the risk of a person with Brugada syndrome experiencing dangerous abnormal heart rhythms.[36] Risk stratification is also sometimes performed using a signal averaged ECG.[5] Ambulatory ECG monitoring, including implantation of a loop recorder, is sometimes used to assess whether dizziness or faints in a person with Brugada syndrome are due to abnormal heart rhythms or other causes such as vasovagal syncope.[37]
.png.webp) Type 1 Brugada ECG pattern (note non-standard lead position, V5 is placed one intercostal space above V1 and V6 is placed one intercostal space above V2)..
Type 1 Brugada ECG pattern (note non-standard lead position, V5 is placed one intercostal space above V1 and V6 is placed one intercostal space above V2)...png.webp) Type 2 Brugada ECG pattern
Type 2 Brugada ECG pattern
Treatment

The main aim when treating people with Brugada syndrome is to reduce the risk of sudden death due to serious abnormal heart rhythms such as ventricular fibrillation or polymorphic ventricular tachycardia.[38] While some with this condition are at high risk of serious heart rhythm disturbances, others are at much lower risk, meaning that some may require more intensive treatment than others.[9] In addition to treating the person who has Brugada syndrome, it is often important to investigate members of their immediate family to see if they too carry the condition.[9]
Lifestyle
The first line of treatment, suitable for all people with Brugada syndrome regardless of their risk of arrhythmias, is lifestyle advice.[9] People should be advised to recognise and avoid things that may increase the risk of serious arrhythmias. These include avoiding excessive alcohol consumption, avoiding certain medications,[12] and treating fever promptly with paracetamol.[9] Although the abnormal heart rhythms seen in Brugada syndrome are generally more likely to occur at rest or even during sleep, some people with Brugada syndrome experience arrhythmias during strenuous exercise. Some physicians may therefore advise people with Brugada syndrome that while gentle exercise is helpful, very strenuous exercise should be avoided.[39][40]
Implantable defibrillator
In people felt to be at higher risk of sudden cardiac death, an implantable cardioverter-defibrillator (ICD) may be recommended.[9] These small devices implanted under the skin continuously monitor the heart rhythm. If the device detects a potentially life-threatening arrhythmia it can give the heart a small electric shock, stunning the heart back into a normal rhythm.[41] An ICD can also function as a pacemaker, preventing abnormally slow heart rates that can also occur in people with Brugada syndrome.
Implanting an ICD is a relatively low-risk procedure and is frequently performed as a day case under local anaesthetic.[41] However, complications such as infection, bleeding or unnecessary shocks can occur, which can sometimes be serious.[42] Because of the small risk associated with implanting an ICD, as well as the cost of the devices, ICDs are not recommended for all people with Brugada syndrome but are instead reserved for people deemed at higher risk of sudden cardiac death.[9]
Medication
Quinidine is an antiarrhythmic drug that may reduce the chance of serious abnormal heart rhythms occurring in some people with Brugada syndrome.[10][43] It is most frequently used in people with Brugada syndrome who have an ICD and have experienced several episodes of life-threatening arrhythmias, but may also be used in people at high risk of arrhythmias but in whom an ICD is not appropriate.[9]
Isoprenaline, a drug that has similarities with adrenaline, can be used in an emergency for people with Brugada syndrome who are having frequent repeated life-threatening arrhythmias, known as an "electrical storm".[9] This drug must be given as a continuous infusion into a vein and therefore is not suitable for long-term use.
Catheter ablation
A further treatment option for people with Brugada syndrome is radiofrequency catheter ablation.[44] In this procedure, wires are passed through a vein in the leg into the heart, or through a small hole underneath the sternum. These wires are used to find the area of the heart responsible for initiating the arrhythmias. The tip of one of these wires is used to make a series of tiny burns, intentionally damaging the area of abnormal heart muscle that has been causing the problem. Current recommendations suggest that this treatment should be reserved for those with Brugada syndrome who have had repeated shocks from an ICD.[9]
Epidemiology
Between 1 and 30 per 10,000 people are affected by Brugada syndrome.[1][2] Although those affected are born with the condition, symptoms typically only begin in adulthood. While the rare cases seen in childhood are equally likely to be male or female, in adulthood symptoms occur more frequently in males than females, potentially due to the higher testosterone levels found in men.[2][45]
Brugada syndrome is more common in people of Asian descent and is the most common cause of sudden death in young males without known underlying cardiac disease in Thailand and Laos.[2][46] In these countries Brugada syndrome is likely to be responsible for many cases of sudden unexpected nocturnal death syndrome (SUNDS). Local names vary – in the Philippines the condition has been known as Bangungut meaning "a scream followed by sudden death during sleep",[46] while in Thailand it was known as Lai Tai, and in Japan Pokkuri.[47][48] Type 1 Brugada ECG patterns are seen more frequently in Asian populations (0–0.36%) than those in Europe (0–0.25%) and the United States (0.03%). Similarly, Type 2 and Type 3 ECG patterns are more prevalent in Asia (0.12–2.23%) than in Europe (0.0–0.6%) or the United States (0.02%).[49]
History
Brugada syndrome is named after the Spanish cardiologists Josep and Pedro Brugada who described the condition in 1992,[11] although the association between the characteristic ECG pattern and sudden cardiac death had been reported in 1989.[50] Brugada syndrome was described as a cause for the sudden unexplained cardiac death syndrome seen in Thai men in 1997.[48] The first genetic mutations affecting the SCN5A gene associated with the syndrome were identified by their brother Ramon Brugada in 1998,[51] with many more mutations affecting at least 19 genes subsequently identified by others.[14] Studies in the 2000s led to competing theories surrounding the mechanisms by which abnormal heart rhythms were generated.[52] Research into Brugada syndrome is ongoing, identifying new genetic variants, exploring mechanisms of arrhythmias, and searching for better treatments.[52]
Society and culture
- A 1992 segment of the television series Unsolved Mysteries profiled the Guamanian Santos family that had lost various members due to heart issues. Surviving members of the family were ultimately diagnosed with Brugada syndrome.[53][54]
- The British television soap opera EastEnders featured a storyline in which one of the characters experienced cardiac arrest due to Brugada syndrome.[55]
- In episode 8 of the 8th season of the TV sitcom Scrubs ("My Lawyer's in Love"), Perry Cox asks a final question of intern Ed which he is unable to answer. He is subsequently fired. One correct answer to his question is Brugada Syndrome, while Long QT syndrome would have also been an acceptable answer.
- In "Transplant", Season 2, Episode 2, a patient of South Asian descent is diagnosed with it.
References
- "Brugada syndrome". Genetics Home Reference. March 2015. Archived from the original on 28 October 2017. Retrieved 28 October 2017.
- Polovina MM, Vukicevic M, Banko B, Lip GY, Potpara TS (October 2017). "Brugada syndrome: A general cardiologist's perspective". European Journal of Internal Medicine. 44: 19–27. doi:10.1016/j.ejim.2017.06.019. PMID 28645806.
- "Brugada Syndrome". NORD (National Organization for Rare Disorders). 2016. Archived from the original on 11 February 2017. Retrieved 28 October 2017.
- "Brugada syndrome". Genetic and Rare Diseases Information Center (GARD) – an NCATS Program. 2017. Archived from the original on 17 October 2017. Retrieved 28 October 2017.
- Sarquella-Brugada G, Campuzano O, Arbelo E, Brugada J, Brugada R (January 2016). "Brugada syndrome: clinical and genetic findings". Genetics in Medicine. 18 (1): 3–12. doi:10.1038/gim.2015.35. PMID 25905440.
- Antzelevitch C, Patocskai B (January 2016). "Brugada Syndrome: Clinical, Genetic, Molecular, Cellular, and Ionic Aspects". Current Problems in Cardiology. 41 (1): 7–57. doi:10.1016/j.cpcardiol.2015.06.002. PMC 4737702. PMID 26671757.
- Doty, Benjamin; Kim, Elaine; Phelps, Jeremiah; Akpunonu, Peter (8 July 2020). "Pathophysiology of Hyperkalemia Presenting as Brugada Pattern on Electrocardiogram (ECG)". The American Journal of Case Reports. 21: e923464. doi:10.12659/AJCR.923464. ISSN 1941-5923. PMC 7370581. PMID 32636355.
- Antzelevitch C, Viskin S (2013). "Brugada Syndrome: Cellular Mechanisms and Approaches to Therapy". In Gussak I, Antzelevitch C, Wilde AA, Powell BD, Ackerman MJ, Shen WK (eds.). Electrical diseases of the heart. Basic foundations and primary electrical diseases. Vol. 1 (2nd ed.). London: Springer. pp. 497–536. ISBN 978-1-4471-4880-7. OCLC 841465583.
- Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, et al. (December 2013). "HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013". Heart Rhythm. 10 (12): 1932–63. doi:10.1016/j.hrthm.2013.05.014. PMID 24011539.
- Belhassen B, Glick A, Viskin S (September 2004). "Efficacy of quinidine in high-risk patients with Brugada syndrome". Circulation. 110 (13): 1731–7. doi:10.1161/01.CIR.0000143159.30585.90. PMID 15381640.
- Brugada P, Brugada J (November 1992). "Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report". Journal of the American College of Cardiology. 20 (6): 1391–6. doi:10.1016/0735-1097(92)90253-J. PMID 1309182.
- Postema PG, Wolpert C, Amin AS, Probst V, Borggrefe M, Roden DM, Priori SG, Tan HL, Hiraoka M, Brugada J, Wilde AA (September 2009). "Drugs and Brugada syndrome patients: review of the literature, recommendations, and an up-to-date website (www.brugadadrugs.org)". Heart Rhythm. 6 (9): 1335–41. doi:10.1016/j.hrthm.2009.07.002. PMC 2779019. PMID 19716089.
- "Brugada Syndrome". National Organization for Rare Disorders. NORD. 2013. Retrieved 28 November 2021.
- Kapplinger JD, Tester DJ, Alders M, Benito B, Berthet M, Brugada J, et al. (January 2010). "An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing". Heart Rhythm. 7 (1): 33–46. doi:10.1016/j.hrthm.2009.09.069. PMC 2822446. PMID 20129283.
- Hedley PL, Jørgensen P, Schlamowitz S, Moolman-Smook J, Kanters JK, Corfield VA, Christiansen M (September 2009). "The genetic basis of Brugada syndrome: a mutation update". Human Mutation. 30 (9): 1256–66. doi:10.1002/humu.21066. PMID 19606473.
- Ciconte G, Monasky MM, Santinelli V, Micaglio E, Vicedomini G, Anastasia L, Negro G, Borrelli V, Giannelli L, Santini F, de Innocentiis C, Rondine R, Locati ET, Bernardini A, Mazza BC, Mecarocci V, Ćalović Ž, Ghiroldi A, D'Imperio S, Benedetti S, Di Resta C, Rivolta I, Casari G, Petretto E, Pappone C (2021). "Brugada syndrome genetics is associated with phenotype severity". Eur Heart J. 42 (11): 1082–1090. doi:10.1093/eurheartj/ehaa942. PMC 7955973. PMID 33221895.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - Pappone C, Ciconte G, Micaglio E, Monasky MM (2021). "Common modulators of Brugada syndrome phenotype do not affect SCN5A prognostic value". Eur Heart J. 42 (13): 1273–1274. doi:10.1093/eurheartj/ehab071. PMC 8014514. PMID 33595071.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - Postema PG, Walsh R, Bezzina CR (2021). "Illuminating the path from genetics to clinical outcome in Brugada syndrome". Eur Heart J. 42 (11): 1091–1093. doi:10.1093/eurheartj/ehaa994. PMC 7955964. PMID 33444429.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - Antzelevitch C (June 2007). "Genetic basis of Brugada syndrome". Heart Rhythm. 4 (6): 756–7. doi:10.1016/j.hrthm.2007.03.015. PMC 1989771. PMID 17556198.
- Belbachir, N; Portero, V; Al Sayed, ZR; Gourraud, JB; Dilasser, F; Jesel, L; Guo, H; Wu, H; Gaborit, N; Guilluy, C; Girardeau, A; Bonnaud, S; Simonet, F; Karakachoff, M; Pattier, S; Scott, C; Burel, S; Marionneau, C; Chariau, C; Gaignerie, A; David, L; Genin, E; Deleuze, JF; Dina, C; Sauzeau, V; Loirand, G; Baró, I; Schott, JJ; Probst, V; Wu, JC; Redon, R; Charpentier, F; Le Scouarnec, S (21 May 2019). "RRAD mutation causes electrical and cytoskeletal defects in cardiomyocytes derived from a familial case of Brugada syndrome". European Heart Journal. 40 (37): 3081–3094. doi:10.1093/eurheartj/ehz308. PMC 6769825. PMID 31114854.
- Garcia-Elias A, Benito B (February 2018). "Ion Channel Disorders and Sudden Cardiac Death". International Journal of Molecular Sciences. 19 (3): 692. doi:10.3390/ijms19030692. PMC 5877553. PMID 29495624.
- Hu D, Barajas-Martínez H, Pfeiffer R, Dezi F, Pfeiffer J, Buch T, Betzenhauser MJ, Belardinelli L, Kahlig KM, Rajamani S, DeAntonio HJ, Myerburg RJ, Ito H, Deshmukh P, Marieb M, Nam GB, Bhatia A, Hasdemir C, Haïssaguerre M, Veltmann C, Schimpf R, Borggrefe M, Viskin S, Antzelevitch C (July 2014). "Mutations in SCN10A are responsible for a large fraction of cases of Brugada syndrome". Journal of the American College of Cardiology. 64 (1): 66–79. doi:10.1016/j.jacc.2014.04.032. PMC 4116276. PMID 24998131.
- Monasky MM, Micaglio E, Vicedomini G, Locati ET, Ciconte G, Giannelli L, Giordano F, Crisà S, Vecchi M, Borrelli V, Ghiroldi A, D'Imperio S, Di Resta C, Benedetti S, Ferrari M, Santinelli V, Anastasia L, Pappone C (2019). "Comparable clinical characteristics in Brugada syndrome patients harboring SCN5A or novel SCN10A variants". Europace. 21 (10): 1550–1558. doi:10.1093/europace/euz186. PMID 31292628. Retrieved 27 April 2021.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - Monasky MM, Micaglio E, Ciconte G, Pappone C (2020). "Brugada Syndrome: Oligogenic or Mendelian Disease?". Int J Mol Sci. 21 (5): 1687. doi:10.3390/ijms21051687. PMC 7084676. PMID 32121523.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - Hoogendijk MG (2012). "Diagnostic dilemmas: overlapping features of brugada syndrome and arrhythmogenic right ventricular cardiomyopathy". Frontiers in Physiology. 3: 144. doi:10.3389/fphys.2012.00144. PMC 3358709. PMID 22654761.
- "KCNT1 protein expression summary - The Human Protein Atlas". www.proteinatlas.org. Retrieved 18 November 2022.
- Kohli, Utkarsh; Ravishankar, Chitra; Nordli, Douglas (December 2020). "Cardiac phenotypic spectrum of KCNT1 mutations". Cardiology in the Young. 30 (12): 1935–1939. doi:10.1017/S1047951120002735. ISSN 1047-9511. PMID 32883383. S2CID 221497594.
- Hasdemir C, Payzin S, Kocabas U, Sahin H, Yildirim N, Alp A, Aydin M, Pfeiffer R, Burashnikov E, Wu Y, Antzelevitch C (July 2015). "High prevalence of concealed Brugada syndrome in patients with atrioventricular nodal reentrant tachycardia". Heart Rhythm. 12 (7): 1584–94. doi:10.1016/j.hrthm.2015.03.015. PMID 25998140.
- Letsas KP, Korantzopoulos P, Efremidis M, Weber R, Lioni L, Bakosis G, Vassilikos VP, Deftereos S, Sideris A, Arentz T (March 2013). "Sinus node disease in subjects with type 1 ECG pattern of Brugada syndrome". Journal of Cardiology. 61 (3): 227–31. doi:10.1016/j.jjcc.2012.12.006. PMID 23403368.
- Wilde AA, Postema PG, Di Diego JM, Viskin S, Morita H, Fish JM, Antzelevitch C (October 2010). "The pathophysiological mechanism underlying Brugada syndrome: depolarization versus repolarization". Journal of Molecular and Cellular Cardiology. 49 (4): 543–53. doi:10.1016/j.yjmcc.2010.07.012. PMC 2932806. PMID 20659475.
- Obeyesekere MN, Klein GJ, Modi S, Leong-Sit P, Gula LJ, Yee R, Skanes AC, Krahn AD (December 2011). "How to perform and interpret provocative testing for the diagnosis of Brugada syndrome, long-QT syndrome, and catecholaminergic polymorphic ventricular tachycardia". Circulation: Arrhythmia and Electrophysiology. 4 (6): 958–64. doi:10.1161/CIRCEP.111.965947. PMID 22203660.
- Wilde, A. a. M.; Antzelevitch, C.; Borggrefe, M.; Brugada, J.; Brugada, R.; Brugada, P.; Corrado, D.; Hauer, R. N. W.; Kass, R. S.; Nademanee, K.; Priori, S. G. (November 2002). "Proposed diagnostic criteria for the Brugada syndrome". European Heart Journal. 23 (21): 1648–1654. doi:10.1053/euhj.2002.3382. ISSN 0195-668X. PMID 12448445.
- Bayés de Luna, Antonio; Brugada, Josep; Baranchuk, Adrian; Borggrefe, Martin; Breithardt, Guenter; Goldwasser, Diego; Lambiase, Pier; Riera, Andrés Pérez; Garcia-Niebla, Javier; Pastore, Carlos; Oreto, Giuseppe (September 2012). "Current electrocardiographic criteria for diagnosis of Brugada pattern: a consensus report". Journal of Electrocardiology. 45 (5): 433–442. doi:10.1016/j.jelectrocard.2012.06.004. ISSN 1532-8430. PMID 22920782.
- Wolpert C, Echternach C, Veltmann C, Antzelevitch C, Thomas GP, Spehl S, Streitner F, Kuschyk J, Schimpf R, Haase KK, Borggrefe M (March 2005). "Intravenous drug challenge using flecainide and ajmaline in patients with Brugada syndrome". Heart Rhythm. 2 (3): 254–60. doi:10.1016/j.hrthm.2004.11.025. PMC 1474213. PMID 15851314.
- Giudicessi JR, Ackerman MJ (January 2013). "Genetic testing in heritable cardiac arrhythmia syndromes: differentiating pathogenic mutations from background genetic noise". Current Opinion in Cardiology. 28 (1): 63–71. doi:10.1097/HCO.0b013e32835b0a41. PMC 3705648. PMID 23128497.
- Sroubek J, Probst V, Mazzanti A, Delise P, Hevia JC, Ohkubo K, et al. (February 2016). "Programmed Ventricular Stimulation for Risk Stratification in the Brugada Syndrome: A Pooled Analysis". Circulation. 133 (7): 622–30. doi:10.1161/CIRCULATIONAHA.115.017885. PMC 4758872. PMID 26797467.
- Kubala M, Aïssou L, Traullé S, Gugenheim AL, Hermida JS (June 2012). "Use of implantable loop recorders in patients with Brugada syndrome and suspected risk of ventricular arrhythmia". Europace. 14 (6): 898–902. doi:10.1093/europace/eur319. PMID 21979995.
- Gourraud JB, Barc J, Thollet A, Le Scouarnec S, Le Marec H, Schott JJ, Redon R, Probst V (2016). "The Brugada Syndrome: A Rare Arrhythmia Disorder with Complex Inheritance". Frontiers in Cardiovascular Medicine. 3: 9. doi:10.3389/fcvm.2016.00009. PMC 4842929. PMID 27200363.
- Mascia G, Arbelo E, Ojeda JH, Solimene F, Brugada R, Brugada J (July 2017). "Brugada Syndrome and Exercise Practice: Current Knowledge, Shortcomings and Open Questions". International Journal of Sports Medicine. 38 (8): 573–581. doi:10.1055/s-0043-107240. PMID 28625016. S2CID 3868956.
- Masrur S, Memon S, Thompson PD (May 2015). "Brugada syndrome, exercise, and exercise testing". Clinical Cardiology. 38 (5): 323–6. doi:10.1002/clc.22386. PMC 6711014. PMID 25955277.
- Rickard J, Wilkoff BL (2016). "Advances in implantable cardioverter defibrillator therapy". Expert Review of Cardiovascular Therapy. 14 (3): 291–9. doi:10.1586/14779072.2016.1131124. PMID 26653411. S2CID 6423194.
- Olde Nordkamp LR, Postema PG, Knops RE, van Dijk N, Limpens J, Wilde AA, de Groot JR (February 2016). "Implantable cardioverter-defibrillator harm in young patients with inherited arrhythmia syndromes: A systematic review and meta-analysis of inappropriate shocks and complications". Heart Rhythm. 13 (2): 443–54. doi:10.1016/j.hrthm.2015.09.010. PMID 26385533.
- Yang F, Hanon S, Lam P, Schweitzer P (April 2009). "Quinidine revisited". The American Journal of Medicine. 122 (4): 317–21. doi:10.1016/j.amjmed.2008.11.019. PMID 19249010.
- Kautzner J, Peichl P (June 2017). "Catheter ablation to prevent sudden cardiac death". International Journal of Cardiology. 237: 29–33. doi:10.1016/j.ijcard.2017.03.135. PMID 28433554.
- Behere SP, Weindling SN (September 2017). "Brugada syndrome in children – Stepping into unchartered territory". Annals of Pediatric Cardiology. 10 (3): 248–258. doi:10.4103/apc.APC_49_17. PMC 5594936. PMID 28928611.
- Brugada J, Brugada P, Brugada R (July 1999). "The syndrome of right bundle branch block ST segment elevation in V1 to V3 and sudden death--the Brugada syndrome". Europace. 1 (3): 156–66. doi:10.1053/eupc.1999.0033. PMID 11225790.
- Nakajima K, Takeichi S, Nakajima Y, Fujita MQ (April 2011). "Pokkuri Death Syndrome; sudden cardiac death cases without coronary atherosclerosis in South Asian young males". Forensic Science International. 207 (1–3): 6–13. doi:10.1016/j.forsciint.2010.10.018. PMID 21084168.
- Nademanee K, Veerakul G, Nimmannit S, Chaowakul V, Bhuripanyo K, Likittanasombat K, Tunsanga K, Kuasirikul S, Malasit P, Tansupasawadikul S, Tatsanavivat P (October 1997). "Arrhythmogenic marker for the sudden unexplained death syndrome in Thai men". Circulation. 96 (8): 2595–600. doi:10.1161/01.CIR.96.8.2595. PMID 9355899.
- Mizusawa Y, Wilde AA (June 2012). "Brugada syndrome". Circulation: Arrhythmia and Electrophysiology. 5 (3): 606–16. doi:10.1161/CIRCEP.111.964577. PMID 22715240.
- Martini B, Nava A, Thiene G, Buja GF, Canciani B, Scognamiglio R, Daliento L, Dalla Volta S (December 1989). "Ventricular fibrillation without apparent heart disease: description of six cases". American Heart Journal. 118 (6): 1203–9. doi:10.1016/0002-8703(89)90011-2. PMID 2589161. S2CID 24418607.
- Chen Q, Kirsch GE, Zhang D, Brugada R, Brugada J, Brugada P, Potenza D, Moya A, Borggrefe M, Breithardt G, Ortiz-Lopez R, Wang Z, Antzelevitch C, O'Brien RE, Schulze-Bahr E, Keating MT, Towbin JA, Wang Q (March 1998). "Genetic basis and molecular mechanism for idiopathic ventricular fibrillation". Nature. 392 (6673): 293–6. Bibcode:1998Natur.392..293C. doi:10.1038/32675. PMID 9521325. S2CID 4315426.
- Brugada P (March 2016). "Brugada syndrome: More than 20 years of scientific excitement". Journal of Cardiology. 67 (3): 215–20. doi:10.1016/j.jjcc.2015.08.009. PMID 26627541.
- "Season 4, Episode: 24." Unsolved Mysteries: Original Robert Stack Episodes. Amazon.com. Cosgrove/Meurer Productions, April 2017. Web. 14 April 2017.
- "Unsolved Mysteries with Robert Stack - Season 4, Episode 24 - Full Episode - YouTube". www.youtube.com. Archived from the original on 11 December 2021.
- "EastEnders: does Stacey's son Arthur have Brugada syndrome?". Radio Times. Retrieved 16 January 2018.
External links
- BrugadaDrugs.org, contains a list of drugs to avoid in people with the Brugada syndrome
- GeneReviews: Brugada syndrome