Adrenocortical carcinoma
Adrenocortical carcinoma (ACC) is an aggressive cancer originating in the cortex (steroid hormone-producing tissue) of the adrenal gland.
| Adrenocortical carcinoma | |
|---|---|
| Other names | Adrenal cortical carcinoma, adrenocorticocarcinoma, adrenal cortical cancer, adrenal cortex cancer |
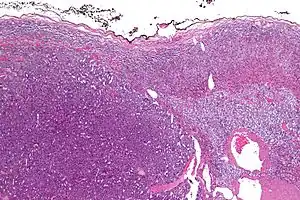 | |
| Micrograph of an adrenocortical carcinoma (left of image – dark blue) and the adrenal cortex it arose from (right-top of image – pink/light blue). Benign adrenal medulla is present (right-middle of image – gray/blue). H&E stain. | |
| Specialty | Oncology |
Adrenocortical carcinoma is remarkable for the many hormonal syndromes that can occur in patients with steroid hormone-producing ("functional") tumors, including Cushing's syndrome, Conn syndrome, virilization, and feminization. Adrenocortical carcinoma has often invaded nearby tissues or metastasized to distant organs at the time of diagnosis, and the overall 5-year survival rate is about 50%.[1]
Adrenocortical carcinoma is a rare tumor, with incidence of one to two per million population annually.[2][3][4][5][6] It has a bimodal distribution by age, with cases clustering in children under 5 and in adults 30–40 years old.[5] The widely used angiotensin-II-responsive steroid-producing cell line H295R was originally isolated from a tumor diagnosed as adrenocortical carcinoma.[7][8]
Signs and symptoms
Adrenocortical carcinoma may present differently in children and adults. Most tumors in children are functional, and virilization is by far the most common presenting symptom(s), followed by Cushing's syndrome and precocious puberty.[5] Among adults presenting with hormonal syndromes, Cushing's syndrome alone is most common, followed by mixed Cushing's and virilization (glucocorticoid and androgen overproduction). Feminization and Conn syndrome (mineralocorticoid excess) occur in less than 10% of cases. Rarely, pheochromocytoma-like hypersecretion of catecholamines has been reported in adrenocortical cancers.[9] Nonfunctional tumors (about 40%, authorities vary) usually present with abdominal or flank pain, varicocele, and renal vein thrombosis[10] or they may be asymptomatic and detected incidentally.[6]
All patients with suspected ACC should be carefully evaluated for signs and symptoms of hormonal syndromes. For Cushing's syndrome (glucocorticoid excess), these include weight gain, muscle wasting, purple lines on the abdomen, a fatty "buffalo hump" on the neck, a "moon-like" face, and thinning, fragile skin. Virilism (androgen excess) is most obvious in women, and may produce excess facial and body hair, acne, enlargement of the clitoris, deepening of the voice, coarsening of facial features, cessation of menstruation. Conn syndrome (mineralcorticoid excess) is marked by high blood pressure, which can result in headache and hypokalemia (low serum potassium, which can in turn produce muscle weakness, confusion, and palpitations), low plasma renin activity, and high serum aldosterone. Feminization (estrogen excess) is most readily noted in men, and includes breast enlargement, decreased libido, and impotence.[5][6][11]
Pathophysiology
The main etiologic factor of ACC is unknown, although families with Li–Fraumeni syndrome, caused by an inherited inactivation mutation in TP53, have increased risk. Several genes have been shown to be recurrently mutated, including TP53, CTNNB1, MEN1, PRKAR1A, RPL22, and DAXX.[12][13] The telomerase gene TERT is often amplified while ZNRF3 and CDKN2A are often homozygously deleted.[13] The genes h19, insulin-like growth factor II (IGF-II), and p57kip2 are important for fetal growth and development. They are located on chromosome 11p. Expression of the h19 gene is markedly reduced in both nonfunctioning and functioning adrenal cortical carcinomas, especially in tumors producing cortisol and aldosterone. Also, a loss occurs of activity of the p57kip2 gene product in virilizing adenomas and adrenal cortical carcinomas. In contrast, IGF-II gene expression has been shown to be high in adrenal cortical carcinomas. Finally, c-myc gene expression is relatively high in neoplasms, and it is often linked to poor prognosis.[14]
Bilateral adrenocortical tumors are less common than unilateral. The majority of bilateral tumours can be distinguished according to size and aspect of the nodules: primary pigmented nodular adrenocortical disease, which can be sporadic or part of Carney complex, and primary bilateral macro nodular adrenal hyperplasia.Metastasis is most commonly to the liver and lung.[15]
Diagnosis
Laboratory findings
Hormonal syndromes should be confirmed with laboratory testing. Laboratory findings in Cushing syndrome include increased serum glucose (blood sugar) and increased urine cortisol. Adrenal virilism is confirmed by the finding of an excess of serum androstenedione and dehydroepiandrosterone. Findings in Conn syndrome include low serum potassium, low plasma renin activity, and high serum aldosterone. Feminization is confirmed with the finding of excess serum estrogen.
Imaging
Radiological studies of the abdomen, such as CT scans and magnetic resonance imaging are useful for identifying the site of the tumor, differentiating it from other diseases, such as adrenocortical adenoma, and determining the extent of invasion of the tumor into surrounding organs and tissues. On CT, it shows heterogeneous appearance due to necrosis, calcifications, and haemorrhage. After contrast injection, it shows peripheral enhancement. Invasion of adjacent structures such as kidney, vena cava, liver, and retroperitoneal lymph nodes are also common.[16]
On MRI, it shows low intensity on T1-weighted images, and high T2 signal with strong heterogeneous contrast enhancement and slow washout. Haemorrhagic areas may show high T1-signal.[16]
Pathology
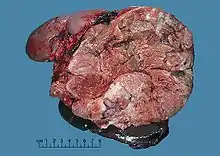
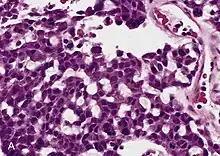
Adrenal tumors are often not biopsied prior to surgery, so diagnosis is confirmed on examination of the surgical specimen by a pathologist. Grossly, ACCs are often large, with a tan-yellow cut surface, and areas of hemorrhage and necrosis. On microscopic examination, the tumor usually displays sheets of atypical cells with some resemblance to the cells of the normal adrenal cortex. The presence of invasion and mitotic activity help differentiate small cancers from adrenocortical adenomas.[9] Several relatively rare variants of ACC include:
- Oncocytic adrenal cortical carcinoma
- Myxoid adrenal cortical carcinoma
- Carcinosarcoma
- Adenosquamous adrenocortical carcinoma
- Clear cell adrenal cortical carcinoma
Differential diagnosis

Differential diagnosis includes:
Adrenocortical carcinomas are most commonly distinguished from adrenocortical adenomas (their benign counterparts) by the Weiss system,[18] as follows:[19]
| Characteristic[19] | Score |
|---|---|
| High nuclear grade (enlarged, oval to lobated, with coarsely granular to hyperchromatic chromatin and easily discernible, prominent nucleoli)[20] | 1 |
| More mitoses than 5/50 high power fields | 1 |
| Atypical mitoses | 1 |
| Eosinophilic cytoplasm in >75% of tumor cells | 1 |
| Diffuse architecture of >33% of tumor | 1 |
| Necrosis | 1 |
| Venous invasion | 1 |
| Sinusoidal invasion (no smooth muscle in wall) | 1 |
| Capsular invasion | 1 |
Total score indicates:[19]
- 0-2: Adrenocortical adenoma
- 3: Undetermined
- 4-9: Adrenocortical carcinoma
Treatment
The only curative treatment is complete surgical excision of the tumor, which can be performed even in the case of invasion into large blood vessels, such as the renal vein or inferior vena cava. The 5-year survival rate after successful surgery is 50–60%, but unfortunately, many patients are not surgical candidates. A 2018 systematic review suggests that laparoscopic retroperotenial adrenalectomy appears to reduce late morbidity, time to oral fluid or food intake and time to ambulation when compared to laparoscopic transperitoneal adrenalectomy, however there is uncertainty about these effects due to very low-quality evidence.[21] For outcomes such as all-cause mortality, early morbidity, socioeconomic effects, and operative and postoperative parameter, the evidence is uncertain about the effects of either interventions over the other.[21]
Radiation therapy and radiofrequency ablation may be used for palliation in patients who are not surgical candidates.[5] Minimally invasive surgical techniques remain controversial due to the absence of long-term data, with a particular concern for rates of recurrence and peritoneal carcinomatosis.
Chemotherapy regimens typically include the drug mitotane, an inhibitor of steroid synthesis, which is toxic to cells of the adrenal cortex,[22][23] as well as standard cytotoxic drugs. A retrospective analysis showed a survival benefit for mitotane in addition to surgery when compared to surgery alone.[24]
The two most common regimens are cisplatin, doxorubicin, etoposide (EDP) + mitotane, and streptozotocin + mitotane. The FIRM-ACT trial demonstrated higher rates of response and longer progression-free survival with EDP + mitotane than with streptozotocin + mitotane.[25]
Prognosis
ACC, generally, carries a poor prognosis,[26] with an overall 5-year survival rate of about 50%.[1] Five-year disease-free survival for a complete resection of a stage I–III ACC is about 30%.[26] The most important prognostic factors are age of the patient and stage of the tumor. Poor prognostic factors include mitotic activity, venous invasion, weight of 50 g or more, diameter of 6.5 cm or more, Ki-67/MIB1 labeling index of 4% or more, and p53 positive.
In its malignancy, adrenocortical carcinoma is unlike most tumours of the adrenal cortex, which are benign (adenomas) and only occasionally cause Cushing's syndrome.
References
- "Adrenal Gland Tumor: Statistics". Cancer.net. 25 June 2012. Retrieved 2020-07-01.
- Wang C, Sun Y, Wu H, Zhao D, Chen J (March 2014). "Distinguishing adrenal cortical carcinomas and adenomas: a study of clinicopathological features and biomarkers". Histopathology. 64 (4): 567–576. doi:10.1111/his.12283. PMC 4282325. PMID 24102952.
- Fassnacht M, Dekkers OM, Else T, Baudin E, Berruti A, de Krijger R, et al. (October 2018). "European Society of Endocrinology Clinical Practice Guidelines on the management of adrenocortical carcinoma in adults, in collaboration with the European Network for the Study of Adrenal Tumors". European Journal of Endocrinology. 179 (4): G1–G46. doi:10.1530/EJE-18-0608. PMID 30299884.
- Fassnacht M, Terzolo M, Allolio B, Baudin E, Haak H, Berruti A, et al. (June 2012). "Combination chemotherapy in advanced adrenocortical carcinoma". The New England Journal of Medicine. 366 (23): 2189–2197. doi:10.1056/NEJMoa1200966. PMID 22551107.
- DeVita VT, Hellman S, Rosenberg SA, eds. (2005). Cancer: principles & practice of oncology. Philadelphia: Lippincott-Raven. ISBN 978-0-7817-4865-0.
- Savarese DM, Nieman LK (2006-08-08). "Clinical presentation and evaluation of adrenocortical tumors". UpToDate Online v. 15.1. UpToDate. Archived from the original on 2007-09-29. Retrieved 2007-06-05.
- Wang T, Rainey WE (March 2012). "Human adrenocortical carcinoma cell lines". Molecular and Cellular Endocrinology. 351 (1): 58–65. doi:10.1016/j.mce.2011.08.041. PMC 3288152. PMID 21924324.
- Gazdar AF, Oie HK, Shackleton CH, Chen TR, Triche TJ, Myers CE, et al. (September 1990). "Establishment and characterization of a human adrenocortical carcinoma cell line that expresses multiple pathways of steroid biosynthesis". Cancer Research. 50 (17): 5488–5496. PMID 2386954.
- Cote R, Suster S, Weiss L (2002). Weidner N (ed.). Modern Surgical Pathology (2 Volume Set). London: W B Saunders. ISBN 978-0-7216-7253-3.
- Cheungpasitporn W, Horne JM, Howarth CB (August 2011). "Adrenocortical carcinoma presenting as varicocele and renal vein thrombosis: a case report". Journal of Medical Case Reports. 5: 337. doi:10.1186/1752-1947-5-337. PMC 3160386. PMID 21806824.
- Kasper DL, Braunwald E, Fauci AS, Hauser SL, Longo DL, Jameson JL. Harrison's Principles of Internal Medicine. New York: McGraw-Hill, 2005. ISBN 0-07-139140-1
- Zheng S, Cherniack AD, Dewal N, Moffitt RA, Danilova L, Murray BA, et al. (May 2016). "Comprehensive Pan-Genomic Characterization of Adrenocortical Carcinoma". Cancer Cell. 29 (5): 723–736. doi:10.1016/j.ccell.2016.04.002. PMC 4864952. PMID 27165744.
- Assié G, Letouzé E, Fassnacht M, Jouinot A, Luscap W, Barreau O, et al. (June 2014). "Integrated genomic characterization of adrenocortical carcinoma". Nature Genetics. 46 (6): 607–612. doi:10.1038/ng.2953. PMID 24747642. S2CID 13089427.
- Kufe D (2000). Benedict RC, Holland JF (eds.). Cancer medicine (5th ed.). Hamilton, Ont: B.C. Decker. ISBN 978-1-55009-113-7. OCLC 156944448.
- James Norman. "Diseases of the Adrenal Cortex: Adrenal Cancer". EndocrineWeb. Updated on: 04/14/16
- Albano D, Agnello F, Midiri F, Pecoraro G, Bruno A, Alongi P, et al. (January 2019). "Imaging features of adrenal masses". Insights into Imaging. 10 (1): 1. doi:10.1186/s13244-019-0688-8. PMC 6349247. PMID 30684056.
- Data and references for pie chart are located at file description page in Wikimedia Commons.
- Wang C, Sun Y, Wu H, Zhao D, Chen J (March 2014). "Distinguishing adrenal cortical carcinomas and adenomas: a study of clinicopathological features and biomarkers". Histopathology. 64 (4): 567–576. doi:10.1111/his.12283. PMC 4282325. PMID 24102952.
- Saygin D, Tabib T, Bittar HE, Valenzi E, Sembrat J, Chan SY, et al. (2015). "Transcriptional profiling of lung cell populations in idiopathic pulmonary arterial hypertension". Pulmonary Circulation. 10 (1): 27–30. doi:10.15605/jafes.030.01.08. PMC 7052475. PMID 32166015.
- Fojo T (26 December 2016). "Adrenocortical Cancer". Retrieved 2020-07-02.
- Arezzo A, Bullano A, Cochetti G, Cirocchi R, Randolph J, Mearini E, et al. (Cochrane Metabolic and Endocrine Disorders Group) (December 2018). "Transperitoneal versus retroperitoneal laparoscopic adrenalectomy for adrenal tumours in adults". The Cochrane Database of Systematic Reviews. 12 (12): CD011668. doi:10.1002/14651858.CD011668.pub2. PMC 6517116. PMID 30595004.
- Fassnacht M, Terzolo M, Allolio B, Baudin E, Haak H, Berruti A, et al. (June 2012). "Combination chemotherapy in advanced adrenocortical carcinoma". The New England Journal of Medicine. 366 (23): 2189–2197. doi:10.1056/NEJMoa1200966. PMID 22551107.
- Brunton LL, Lazo JS, Parker KL, eds. (2006). Goodman & Gilman's The Pharmacological Basis of Therapeutics (11th ed.). United States of America: The McGraw-Hill Companies, Inc. ISBN 978-0-07-142280-2.
- Terzolo M, Angeli A, Fassnacht M, Daffara F, Tauchmanova L, Conton PA, et al. (June 2007). "Adjuvant mitotane treatment for adrenocortical carcinoma". The New England Journal of Medicine. 356 (23): 2372–2380. doi:10.1056/NEJMoa063360. hdl:2318/37317. PMID 17554118.
- Fassnacht M, Terzolo M, Allolio B, Baudin E, Haak H, Berruti A, et al. (June 2012). "Combination chemotherapy in advanced adrenocortical carcinoma". The New England Journal of Medicine. 366 (23): 2189–2197. doi:10.1056/NEJMoa1200966. hdl:2318/102217. PMID 22551107.
- Allolio B, Fassnacht M (June 2006). "Clinical review: Adrenocortical carcinoma: clinical update". The Journal of Clinical Endocrinology and Metabolism. 91 (6): 2027–2037. doi:10.1210/jc.2005-2639. PMID 16551738. Free Full Text.
{kind=link}