Angiocentric glioma
Angiocentric glioma (AG) refers to a rare neuroepithelial tumor when the superficial brain malignant cells enclose the brain vessels, commonly found in children and young adults. Initially identified in 2005 by Wang and his team from the University of Texas, AG was classified as Grade I by 2007 WHO Classification of Tumors of the Central Nervous System due to its benign clinical behavior, low proliferation index, and curative properties.[2] AG primarily affects children and young adults at an average initial diagnosis age of 16 years old. Over 85% AG patients experience intractable seizures since childhood, especially partial epilepsy.[3]
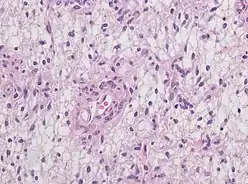
Due to its short history of 15 years, the rarity of occurrence, and a lack of sufficient clinical trials, AG remains elusive on understanding symptoms, treatments, and long-term follow-up. Till now, scientists and researchers have not found the exact etiology, definitive pathological tests for identification, and the effect of radiation or chemotherapy on this rare indolent glioma. Yet, a series of suspected causes are under discussion, including the possible MYB-QKI protein fusion theory on AG etiology. Currently, the standard diagnostic tools are MRI (Magnetic Resonance Imaging) and Computed Tomography scan (CT scan). In terms of therapy, patients often undergo subtotal or total resection to remove the problematic lesion and have a relatively high likelihood of curing the disease. However, they still require more extended follow-up periods after surgery for monitoring tumor recurrence and assuring seizure-free.
History
Prior to AG's initiation, the most frequent and best-known types of rare glioneuronal tumors in the brain were dysembryoplastic neuroepithelial tumors and gangliogliomas.[4] In 2002, Wang and his group from the University of Texas recognised a novel neurological pattern in three cerebral hemispheric tumor cases and first published this rare subtype of AG tumor to the American Association of Neuropathologists.[5] In 2005, Lellouch-Tubiana and his team analyzed 204 epilepsy surgery specimens histologically from patients aged 2 to 14 and discovered three very similar cases shown by Wang et al. in terms of pathological appearance and MRI results.[4] AG manifests a distinct architectural neuron pattern and morphologic features, which were subsequently characterized as a new clinicopathologic entity named "angiocentric neuroepithelial tumors" within the spectrum of children glioneuronal tumors.[4] Simultaneously in the same month in 2005, Wang et al. also published their further discoveries of five AG cases and indicated the specified histologic, immunohistochemical, and ultrastructural properties of this seizure-producing low growing tumor.[5] In 2007, the Fourth Edition of the World Health Organization Classification of Tumors of the Central Nervous System recognized AG as a new clinicopathologic entity and designated it as an epilepsy-associated Grade I tumor with monomorphous bipolar cells and a unique perivascular growth arrangement in the category of "other neuroepithelial tumors".[2] In 2016, WHO made a few amendments in the tumor classification parameter and classified AG, choroid glioma of the third ventricle, and astroblastoma as Grade I "Other Gliomas" after revision.[6]
Signs and symptoms
Patients usually experience a history of intractable seizures at 3 to 14 years old.[7] The seizure severity often depends on the tumor locations rather than its size, as superficial lesions located in the frontal and temporal lobes often trigger a higher epileptogenic possibility than the deeper tumors.[8] Common symptoms also include headache, vision impairment, dizziness, otalgia, speech arrest, ataxia, and paresis, predominantly in children and young adults.[9] The symptoms of low-grade, slow-growing gliomas are more epileptogenic, whereas those high-grade gliomas manifest symptoms related to increased intracranial pressure.[10]
Causes
Many studies on AG have prioritized neuroradiological features, clinical highlights, pathophysiology, and surgical treatment of this rare disease while lacking discussion on its causes. AG's cytogenesis has remained elusive since 2005, and researchers only proposed several possible mechanisms for this low-grade child brain tumor.
As early as 2005, the first published research on AG postulated that ependymoma and variants of astroblastoma might contribute to the AG tumor formation.[5] In the same year, another group of researchers suggested AG possibly derives from the neoplastic transformation of radial glial cells during neuronal migration as they present a similar proliferation pattern.[4] However, these two publications have not provided further experiments to prove their rationales.
MYB-QKI Protein Fusion
In 2016, Bandopadhayay and his research team proposed a potential genetic abnormality attributor for AG formation. According to their proposal, gene fusions that activate the mitogen-associated protein kinase (MAPK) pathway can induce AG malignant cells.[11] After completing combined genomic analysis of whole-genome sequencing and RNA sequencing data from 172 paediatric low-grade gliomas (PLGG) subtypes, they recognized protein fusion of the MYB and QKI gene in most AG profiles, thus hypothesizing it to be the tumor-causing driver. MYB is a proto-oncogene, whereas QKI is a tumor-suppressor gene. The abnormal fusion of the two may impose a malignant effect on neurologic cells as the rearrangement of QKI and MYB can prompt an increased expression of in-frame MYB-QKI fusion protein than the normal paediatric cortical brain. The research group proved that this protein alternation could be the critical contributor to AG oncogenicity since mice showed an enhanced cell proliferation rate after intracranial injection in experiments. The same article also pointed out that MYB-QKI protein facilitates the growth of vessels around the glioma.
Low recurrence after surgery
AG often behaves as a low-grade indolent neoplasm and is curative after surgical resection. Researchers proposed that since AG does not acquire mutations of isocitrate dehydrogenase-1, making it have a lower recurrence potential after surgery compared to WHO grade II and III diffuse gliomas and secondary glioblastomas.[3]
Diagnosis
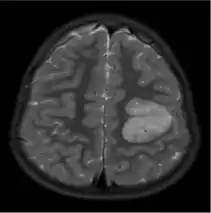
The neurological features of AG tumors are visible via CT scanning or MRI. A clear indication of AG may appear as well-delineated, solid, T2-hyperintense, non-enhancing cortical lesions located in the temporal or frontal lobes in MRI.[1] Another diagnostic trait is a stalk-like extension to adjacent brain ventricles.[13] These traits are similar to low-grade gliomas from a radiological perspective.
The results from CT scanning and MRI are different in terms of clarity and effectiveness of diagnosis. AG displays an expansive non-enhancing cortical tumor in CT scanning, whereas MRI shows a relatively clearer appearance of AG and the tumors appear to be infiltrative, well-defined, and hypointense with T1 lesion.[4] T2/FLAIR lesions indicate AG as a tumor tissue with some extension toward the ventricles along vessels.[4] The possibility of cystic-appearing areas exists as well. In some cases, MRI results show an increase in ribbon-like signal on T1W1 lesions.[4] The clear radiographic outcomes of MRI makes it the more widely used option in the diagnosis of AG.[4]
Nevertheless, precise diagnosis of AG from other phenotypically similar gliomas (such as astroblastoma or ganglioglioma) is a challenge merely based on MRI or CT scanning.[14] The main difference between AG and ganglioglioma could be only AG shows enhancement over time. Compared to AG, astroblastoma often has a discrete border in epithelioid cells and shows vascular sclerosis symptoms.[15]
For further confirmation, the clinicians require biopsy and immunohistochemical staining of the resected tumor after surgery. The infiltrative AG cells display positive results for several immunostainings, especially the glial fibrillary acidic protein (GFAP) and epithelial membrane antigen (EMA).[1] Clinicians also observe a specific dot-like pattern from the stained EMA photomicrograph.[3] Other specific AG immunohistochemical tests include Ki-67 proliferative marker, neurospecific nucleoprotein (NeuN), protein 53, synaptophysin (Syn), oligodendrocyte transcription factor-2 (Olig-2) and creatine kinase (CK).[3] In the 2016 WHO classification of CNS tumors, AG is characterised as GFAP-positive, NeuN-positive and low Ki-67 proliferative rate with a perivascular growth pattern.[13]
Treatment
The definitive treatment for this low-grade entity is surgical resection. Given its rarity and short history, clinicians have not found other better treatment options for AG.[14] Nearly all AG patients either choose subtotal or total resection to remove the tumor tissue in the brain surgically. A total resection manages to regress epileptogenic growth and cure this brain neoplasm.[14] Subtotal resection shows a comparably higher recurrence rate and tumor progression with limited control of seizures.[14] The overall outcome of tumoral resection is highly ideal with a considerably favorable prognosis.[16] Additionally, the effectiveness of chemotherapy or radiation is yet to be known as these aggressive treatments are considered inappropriate for low-grade AG.[17] It is also rare to aid the treatment with extra radiotherapy.[14]
Epidemiology
By June 2020, the reported AG cases have reached 108 since the initial report from Wang et al. Within the reported cases, it happens mostly in children and young adults.[3] The average age of initial diagnosis is 16 year-old, and the prevalence in males to females is in a ratio of 1.5 to 1.[3] The initial diagnosis range varies from the age of 1.5 to 83, with a median of 13.[3]
102 out of 108 reported cases had AG tumors in a supratentorial location under the cerebral cortex (94.4%), and 88 out of 108 were found in a single lobe (81.5%).[3] 46 cases were in the left lobe of the brain and 43 in the right lobe.[3] The most common location where AG starts to grow is the temporal lobe with 39% of reported cases, followed by parietal (30%) and frontal lobes (15%).[9] The less common growth site is the thalamus, with only 1% of reported cases, and only six cases of brainstem lesions have been reported.[8]
94 out of 108 (87%) patients chose to conduct a certain degree of surgical removal: 61 patients took gross total resection (GTR, 64.9%), and 26 adopted subtotal resection (STR, 27.7%).[3] The resection outcome was highly ideal, with 93.1% of the patient completely free of seizure during the follow-up period.[3] The remaining six patients who chose to conduct STR experienced some degrees of reoccurred symptoms.[3] This indicates that AG has a favourable prognosis with a comparatively low mortality rate and recurrence rate.
References
- Shakur SF, McGirt MJ, Johnson MW, Burger PC, Ahn E, Carson BS, Jallo GI (March 2009). "Angiocentric glioma: a case series". Journal of Neurosurgery. Pediatrics. 3 (3): 197–202. doi:10.3171/2008.11.PEDS0858. PMC 2675755. PMID 19338465.
- Brat DJ, Scheithauer BW, Fuller GN, Tihan T (July 2007). "Newly codified glial neoplasms of the 2007 WHO Classification of Tumours of the Central Nervous System: angiocentric glioma, pilomyxoid astrocytoma and pituicytoma". Brain Pathology. 17 (3): 319–324. doi:10.1111/j.1750-3639.2007.00082.x. PMC 8095654. PMID 17598825.
- Han G, Zhang J, Ma Y, Gui Q, Yin S (August 2020). "Clinical characteristics, treatment and prognosis of angiocentric glioma". Oncology Letters. 20 (2): 1641–1648. doi:10.3892/ol.2020.11723. PMC 7377082. PMID 32724405.
- Lellouch-Tubiana A, Boddaert N, Bourgeois M, Fohlen M, Jouvet A, Delalande O, et al. (October 2005). "Angiocentric neuroepithelial tumor (ANET): a new epilepsy-related clinicopathological entity with distinctive MRI". Brain Pathology. 15 (4): 281–286. doi:10.1111/j.1750-3639.2005.tb00112.x. PMC 8095937. PMID 16389940.
- Wang M, Tihan T, Rojiani AM, Bodhireddy SR, Prayson RA, Iacuone JJ, et al. (October 2005). "Monomorphous angiocentric glioma: a distinctive epileptogenic neoplasm with features of infiltrating astrocytoma and ependymoma". Journal of Neuropathology and Experimental Neurology. 64 (10): 875–881. doi:10.1097/01.jnen.0000182981.02355.10. PMID 16215459. S2CID 15439580.
- Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. (June 2016). "The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary". Acta Neuropathologica. 131 (6): 803–820. doi:10.1007/s00401-016-1545-1. PMID 27157931. S2CID 3345100.
- Preusser M, Hoischen A, Novak K, Czech T, Prayer D, Hainfellner JA, et al. (November 2007). "Angiocentric glioma: report of clinico-pathologic and genetic findings in 8 cases". The American Journal of Surgical Pathology. 31 (11): 1709–1718. doi:10.1097/PAS.0b013e31804a7ebb. PMID 18059228. S2CID 41723081.
- da Silva JF, de Souza Machado GH, Pedro MK, Vosgerau R, Hunhevicz SC, Ramina R (Dec 2019). "Angiocentric glioma: Literature review and first case in Brazil". Interdisciplinary Neurosurgery. 18: 100508. doi:10.1016/j.inat.2019.100508. ISSN 2214-7519. S2CID 196565334.
- Alexandru D, Haghighi B, Muhonen MG (2013-03-01). "The treatment of angiocentric glioma: case report and literature review". The Permanente Journal. 17 (1): e100–e102. doi:10.7812/tpp/12-060. PMC 3627792. PMID 23596378.
- Ampie L, Choy W, DiDomenico JD, Lamano JB, Williams CK, Kesavabhotla K, et al. (June 2016). "Clinical attributes and surgical outcomes of angiocentric gliomas". Journal of Clinical Neuroscience. 28: 117–122. doi:10.1016/j.jocn.2015.11.015. PMID 26778052. S2CID 1420908.
- Bandopadhayay P, Ramkissoon LA, Jain P, Bergthold G, Wala J, Zeid R, et al. (March 2016). "MYB-QKI rearrangements in angiocentric glioma drive tumorigenicity through a tripartite mechanism". Nature Genetics. 48 (3): 273–282. doi:10.1038/ng.3500. PMC 4767685. PMID 26829751.
- Kumar M, Ramakrishnaiah R, Samant R (2013). "Angiocentric glioma, a recently added WHO grade-I tumor". Radiology Case Reports. 8 (4): 782. doi:10.2484/rcr.v8i4.782. PMC 4899553. PMID 27330646.
- Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (2016). WHO classification of tumours of the central nervous system (Revised 4th ed.). Lyon: World Health Organization, International Agency for Research on Cancer. ISBN 978-92-832-4492-9. OCLC 951745876.
- Gatto L, Franceschi E, Nunno VD, Tomasello C, Bartolini S, Brandes AA (2020-07-03). "Glioneuronal tumors: clinicopathological findings and treatment options". Future Neurology. 15 (3): FNL47. doi:10.2217/fnl-2020-0003. ISSN 1479-6708. S2CID 225450256.
- Zhou L, Burns DK, Cai C, eds. (2020). A Case-Based Guide to Neuromuscular Pathology. Springer International Publishing. doi:10.1007/978-3-030-25682-1. ISBN 978-3-030-25682-1. S2CID 204886503.
- Osborn AG, Salzman KL, Thurnher MM, Rees JH, Castillo M (May 2012). "The new World Health Organization Classification of Central Nervous System Tumors: what can the neuroradiologist really say?". AJNR. American Journal of Neuroradiology. 33 (5): 795–802. doi:10.3174/ajnr.A2583. PMC 7968804. PMID 21835942.
- Mayor S (2005-05-05). "Aggressive treatment of low grade prostate cancer is unnecessary". BMJ. 330 (7499): 1042.2. doi:10.1136/bmj.330.7499.1042-a. ISSN 0959-8138. PMC 557253.