Animal model of schizophrenia
Research into the mental disorder of schizophrenia, involves multiple animal models as a tool, including in the preclinical stage of drug development.

Several models simulate schizophrenia defects. These fit into four basic categories: pharmacological models, developmental models, lesion models, and genetic models. Historically, pharmacological, or drug-induced models were the most widely used. These involve the manipulation of various neurotransmitter systems, including dopamine, glutamate, serotonin, and GABA. Lesion models, in which an area of an animal's brain is damaged, arose from theories that schizophrenia involves neurodegeneration, and that problems during neurodevelopment cause the disease. Traditionally, rodent models of schizophrenia mostly targeted symptoms analogous to the positive symptoms of schizophrenia, with some models also having symptoms similar to the negative symptoms. Recent developments in schizophrenia research, however, have targeted cognitive symptoms as some of the most debilitating and influential in patients' daily lives, and thus have become a larger target in animal models of schizophrenia.[1] Animals used as models for schizophrenia include rats, mice, and primates.
Uses and limitations
The modelling of schizophrenia in animals can range from attempts to imitate the full extent of symptoms found in schizophrenia, to more specific modelling which investigate the efficacy of antipsychotic drugs. Each extreme has its limitations, with whole-syndrome modeling often failing due to the complexity and heterogeneous nature of schizophrenia, as well as difficulty translating human specific diagnostic criteria such as disorganized speech to animals. Antipsychotic-specific modelling faces similar issues, one of which is that it is not useful for discovering drugs with unique mechanisms of action, while traditional medications for schizophrenia have generalized effects (blocking of dopamine receptors) that make it difficult to attribute outcomes to schizophrenia specifically. Developing models based on a particular sign or symptom of schizophrenia has thus become a more common approach. This approach has the advantage that the results are more likely to be valid across the species boundary.[2]
In order for an animal model to be useful in developing treatments, results from the animal model must translate into results in the patient with schizophrenia, this is called the validity of the model.[3] Criteria for assessing the validity of animal models of schizophrenia include face validity, construct validity, and predictive validity.[3][4] While no animal model can fully encompass all aspects of schizophrenia, progress has been made in using animals to model schizophrenia and its relationship to other mental disorders, such as addiction.[5]
Phenotype
The validity of an animal model of schizophrenia can be measured using several behavioural, cellular and anatomical traits (the phenotype of the model).[2]
Behavioural traits
- Prepulse inhibition
- Prepulse inhibition (PPI) is the inhibition of the response to a startling stimulus when the stimulus is preceded by a weaker stimulus. Abnormalities in PPI are seen in patients with schizophrenia.[2][6]
- Latent inhibition
- Anomalies in latent inhibition have been linked with schizophrenia in acute events. in latent inhibition, frequent presentation of a stimulus reduces the rate at which the stimulus produces meaning.[2]
- Paired click gating
- In Paired-click gating or P50 gating, a click which quickly follows a previous click produces a reduced response. This gating is reduced in schizophrenia patients relative to normal subjects.[2]
- Social withdrawal
- One of the negative symptoms of schizophrenia is social withdrawal, this has been modelled by socially isolating animals.[2]
- Locomotor traits
- Some locomotor changes, including stereotypy, are used to test the validity of animal models. Locomotor anomalies are often found in patients with schizophrenia.[2]
- Context Processing
- One cognitive symptom of schizophrenia is deficits in context processing. Animal models of schizophrenia have shown some validity in replicating a schizophrenia specific pattern of context processing deficits[1]
- Working Memory
- Deficits in working memory are a core deficit in schizophrenia patients, with comparable deficits being used as a measure of validity in animal models.[7]
Other traits
- Cortical volume
- Reduced volume of the prefrontal cortex is an anatomical trait of schizophrenia patients which is used to test the validity of animal models.[2]
- NMDA receptor gene expression
- Variations in the expression of the NR2B and NR1 subunits of the NMDA receptor are used to test developmental animal models.[2]
- Dopamine release
- Neuroimaging studies on patients with schizophrenia have found increased dopamine release after treatment with amphetamine relative to normal controls.[2]
Pharmacological models
Dopamine
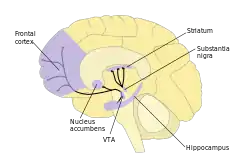
In the dopamine hypothesis of schizophrenia, schizophrenia was hypothesised to be caused by disturbed dopamine neurotransmission. Dopamine is a monoamine neurotransmitter which is involved in other diseases, such as Parkinson's disease. There is evidence for increased activity of the mesolimbic pathway, a dopaminergic pathway, in schizophrenia patients. This comes from the discovery of increased L-DOPA decarboxylase levels in the brains of these patients. L-DOPA decarboxylase is an enzyme which converts L-DOPA to dopamine by removing a carboxyl group.[8] Animal models were first produced for schizophrenia by altering the dopaminergic system using drugs.[9]
Persistent treatment of rodents with amphetamine models show symptoms of schizophrenia including hyperactivity, enduring prepulse inhibition abnormalities, and cognitive abnormalities associated with the prefrontal cortex including attention deficits. Neither negative symptoms, such as problems with social interaction, or hippocampus-related deficits are observed in amphetamine rodent models. The antipsychotics clozapine and haloperidol reverse the effects of amphetamine on attention in rats.[9]
Glutamate
Glutamate is the most abundant excitatory neurotransmitter in vertebrate nervous systems. Evidence for the involvement of glutamate in schizophrenia includes analogous symptoms which are produced by glutamate NMDA receptor antagonists such as phencyclidine (PCP) and ketamine. PCP is a non-competitive NMDA receptor antagonist which produces hallucinations and delusions in normal subjects. In rat models, disturbed cognition, deficits in social interaction, locomotor anomalies, and prepulse inhibition deficits are seen on acute administration of PCP. Evidence that persistent PCP use and abuse in humans results in lasting deficits beyond the period of treatment has led to the suggestion that this regime in rodents may be a more accurate model of schizophrenia than acute administration. A number of protocols for chronic PCP animal models have been developed, with different effects. The effects of some, but not all protocols can be reversed by treatment with antipsychotics. In a primate model, PCP was found to induce cognitive impairments which were reversed with clozapine.[9]
Serotonin
Serotonin is a monoamine neurotransmitter which has been associated with schizophrenia. The psychedelic drug classes indoleamines and phenethylamines can affect serotoninergic 5-HT2A receptors. LSD, an indoleamine, affects startle habituation and prepulse inhibition of startle, which are indicators of human schizophrenia.[8]
GABA
Gamma-Aminobutyric acid (GABA) is a major inhibitory neurotransmitter. The GABAergic system may be involved in schizophrenia due to its interactions with the dopaminergic system. Picrotoxin, an antagonist for the GABAA receptor, produces prepulse inhibition of startle in rats. Haloperidol, an antipsychotic drug, reduces this effect.[8]
Lesions
Studies into the neurodevelopmental and neurodegenerative aspects of schizophrenia have led to the use of lesion models to investigate these aspects. A lesion is damage to an area of tissue by any cause. The evidence for the neurodegenerative theory is a reduction in the volume of the cerebral cortex and an increase in the volumes of the ventricles (cavities in the brain containing cerebrospinal fluid) associated with schizophrenia. Most neurodegenerative diseases produce increased levels of glial cells such as astrocytes, this is not found in schizophrenia. The evidence in favour of the neurodevelopmental theory includes the connection of some physical abnormalities with schizophrenia.[8]
Brain regions used in lesion models of schizophrenia include the prefrontal cortex, the hippocampal formation, and the thalamus. In rat models, lesions of the prefrontal cortex have produced increased and protracted response to stress and a lower prepulse inhibition of startle when treated with apomorphine.[8]
Neonatal ventral hippocampal lesion
Neonatal lesions of the ventral part of the hippocampus in rats (NVHL rats) is a widely studied developmental animal model of schizophrenia. NVHL rats mimic many of the symptoms of schizophrenia in detail.[10] The behavioural deficits caused by NVHL are seen after puberty and include aggression and social interaction abnormalities. The precise effects of the lesion depend on the day on which it is administered.[9]
Developmental models
There is evidence from epidemiological studies that environmental factors during gestation or around childbirth can increase the probability of someone developing schizophrenia.[9]
Methylazoxymethanol acetate
Methylazoxymethanol acetate (MAM) is used during gestation to affect aspects of neural development. MAM selectively targets neuroblasts in the central nervous system. As neuroblasts are cells which become neurons, interfering with them using MAM inhibits the areas of the brain which are developing most quickly. The effects of MAM therefore depend on the stage of development at which it is administered, or the gestational age of the subject. In rat studies, administration of MAM at day 17 of gestation (GD17) results in several cognitive and anatomical changes which are common to schizophrenia patients. The thickness of the hippocampus and the thalamus are reduced, the locomotor effects of amphetamines and the spontaneous firing rate of dopominergic neurons in the ventral tegmental area are increased, and defects in working spatial memory are observed.[9][11]
Social isolation
Rats have a specific social organization within colonies. In social isolation models, pups which are placed in separate cages after being weaned show behavioural changes as adults and altered neural development. These changes remain after being re-introduced into the colony in adulthood. The behavioural deficits caused include neophobia, a larger response to new stimulus, locomotor hyperactivity, and increased aggression. Social isolation rats' inability to habituate to new environments may be caused by an increased mesolimbic dopaminergic activity.[9]
Genetic models
Studies involving twins have shown that schizophrenia is a heritable disease. While no one gene is responsible for the disease, a large number of possible genes have been identified. Genetic animal models of schizophrenia often involve knockout mice, genetically modified mice where one or more of these genes is removed or disrupted.[9]
DISC1
Disrupted in schizophrenia 1 (DISC1) was one of the first genes discovered to be involved in schizophrenia. As of 2011, seven different strains of DISC1 mouse models had been developed. As in schizophrenia patients, DISC1 mice have an increased lateral ventricle size, reduced cortical size, changes to the hippocampus, and changes to prepulse inhibition of startle which are reversed on treatment with haloperidol and clozapine.[9]
One DISC1 mouse model is induced by the mutagenic chemical ENU. ENU introduces missense point mutations; screening for mutations in a particular exon of DISC1 can produce mouse models with schizophrenia-like behavioural deficits.[12]
Neuregulin 1 and associated genes
The gene NRG1 codes for neuregulin 1, a growth factor which is crucial to the development of the nervous system, and to neurotransmission and formation of synapses in adults. NRG1 and the gene for the receptor to which neuregulin 1 binds, ERBB4, have been tested as possible animal models of schizophrenia. While mice which have two copies of (are homozygous for) a knocked out version of NRG1 do not survive, viable animal models have been developed using heterozygous or partial knockout. One such mouse model is the heterozygous removal of the EGF-like domain on neuregulin 1, these models are called Nrg1(ΔEGF)+/− mice. Nrg1(ΔEGF)+/− mice have been shown to have social interaction problems, reduced prepulse of inhibition and larger spontaneous locomotion. Other neuregulin 1 models include the heterozygous removal of the transmembrane domain (Nrg1(ΔTM)+/− mice) and the immunoglobulin domain (Nrg1(ΔIg)+/− mice). Nrg1(ΔTM)+/− mice display hyperactivity in various conditions, an effect which is reduced by the atypical antipsychotic clozapine.[9]
Dysbindin
Dysbindin is a protein coded for by the gene DTNBP1, which has been linked to schizophrenia.[3] Dysbindin may be involved in the changes to neurotransmission observed in patients with schizophrenia. According to SA Jones et al, DTNBP1 is "currently thought to be one of the most promising candidate genes for schizophrenia susceptibility".[9] One naturally occurring animal model involving dysbindin, the sdy (sandy) mouse, has a number of anatomical changes compared to normal mice, including changes to the hippocampus. Sdy mice have homozygous mutations to DTNBP1, and lack the ability to produce dysbindin, heterozygous mutants can be produced by crossing sdy mice with a strain of normal mice.[9]
Reelin
Reelin is a protein which is involved in synaptic plasticity and synaptogenesis in the brain. In the frontal cortices, cerebellums, and hippocampi of schizophrenia patients, the amount of the protein and its messenger RNA are reduced. Knockout mice in which the reelin gene is disrupted are called reeler mice. In homozygous reeler mice, extreme changes in gait and other behavioural anomalies are seen (illustrated in video); these changes go beyond those associated with schizophrenia.[9]
Heterozygous reeler mice display lower social dominance in some tests, but other social deficits seen in schizophrenia are absent.[13] Reeler mice also have schizophrenia-like anatomical defects in the frontal cortex, but have few cognitive defects which are associated with that area and found in schizophrenia. Tests using the Morris water maze have found that reeler mice do not have the abnormalities in spatial reference memory which are found in patients with schizophrenia.[9]
Other genetic models
- G72/G30
- The G72/G30 gene complex is a possible risk factor for schizophrenia; male animal mutants have less aggressive behaviours and have olfactory abnormalities.[13]
- PPP3CC
- PPP3CC is a gene in which mutations are risk factors for schizophrenia; knockout animal models have social deficits.[13]
16p11.2 Duplication
Micro-duplications of a 600kb region of chromosome 16p11.2 have been associated with a significantly increased risk of schizophrenia.[14] This is a region that is conserved across several species, including mice and rats. This has made it a popular target site for current research into rodent models of schizophrenia.
22q11.2 Deletion
A deletion in chromosome 22q11.2 is the strongest known genetic risk associated with schizophrenia, with 25% of individuals with this deletion ultimately testing positive for schizophrenia.[15]
References
- Blackman, Rachael K.; Macdonald, Angus W.; Chafee, Matthew V. (October 2013). "Effects of ketamine on context-processing performance in monkeys: a new animal model of cognitive deficits in schizophrenia". Neuropsychopharmacology. 38 (11): 2090–2100. doi:10.1038/npp.2013.118. ISSN 1740-634X. PMC 3773669. PMID 23660706.
- Geyer, MA; Moghaddam, B (2002). "Chapter 50: Animal Models Relevant To Schizophrenia Disorders". Neuropsychopharmacology : the fifth generation of progress. American College of Neuropsychopharmacology. ISBN 0781728371. Archived from the original on 2013-10-20. Retrieved 2012-06-11.
- Wilson, C; Terry AV, Jr (Jul 2010). "Neurodevelopmental animal models of schizophrenia: role in novel drug discovery and development". Clinical Schizophrenia & Related Psychoses. 4 (2): 124–37. doi:10.3371/CSRP.4.2.4. PMC 4400734. PMID 20643635.
- Young, JW; Zhou, X; Geyer, MA (2010). "Animal Models of Schizophrenia". Behavioral neurobiology of schizophrenia and its treatment. Heidelberg: Springer. pp. 391–433. ISBN 9783642137167.
- Menne, Victoria; Chesworth, Rose (2020-01-16). "Schizophrenia and drug addiction comorbidity: recent advances in our understanding of behavioural susceptibility and neural mechanisms". Neuroanatomy and Behaviour. 2: e10. doi:10.35430/nab.2020.e10. ISSN 2652-1768.
- Wan, L; Thomas, Z; Pisipati, S; Jarvis, SP; Boutros, NN (April 2017). "Inhibitory deficits in prepulse inhibition, sensory gating, and antisaccade eye movement in schizotypy". International Journal of Psychophysiology. 114: 47–54. doi:10.1016/j.ijpsycho.2017.02.003. PMID 28189549.
- Castner, Stacy A.; Goldman-Rakic, Patricia S.; Williams, Graham V. (June 2004). "Animal models of working memory: insights for targeting cognitive dysfunction in schizophrenia". Psychopharmacology. 174 (1): 111–125. doi:10.1007/s00213-003-1710-9. ISSN 0033-3158. PMID 15205882. S2CID 22685001.
- Marcotte, ER; Pearson, DM; Srivastava, LK (November 2001). "Animal models of schizophrenia: a critical review". Journal of Psychiatry & Neuroscience. 26 (5): 395–410. PMC 167198. PMID 11762207.
- Jones, CA; Watson, DJG; Fone, KCF (1 October 2011). "Animal models of schizophrenia". British Journal of Pharmacology. 164 (4): 1162–1194. doi:10.1111/j.1476-5381.2011.01386.x. PMC 3229756. PMID 21449915.
- O'Donnell, P (editor) (2011). Animal models of schizophrenia and related disorders. New York: Humana Press. ISBN 9781617791567.
{{cite book}}:|last=has generic name (help) - Hradetzky, Eva; et al. (28 September 2011). "The Methylazoxymethanol Acetate (MAM-E17) Rat Model: Molecular and Functional Effects in the Hippocampus". Neuropsychopharmacology. 37 (2): 364–377. doi:10.1038/npp.2011.219. PMC 3242314. PMID 21956444.
- Sawa, Akira, ed. (2009). Genetic models of schizophrenia (1st ed.). Amsterdam: Elsevier Science. ISBN 9780444534309.
- O'Tuathaigh, CM; Kirby, BP; Moran, PM; Waddington, JL (Mar 2010). "Mutant mouse models: genotype–phenotype relationships to negative symptoms in schizophrenia". Schizophrenia Bulletin. 36 (2): 271–88. doi:10.1093/schbul/sbp125. PMC 2833123. PMID 19934211.
- McCarthy, Shane E.; Makarov, Vladimir; Kirov, George; Addington, Anjene M.; McClellan, Jon; Yoon, Seungtai; Perkins, Diana O.; Dickel, Diane E.; Kusenda, Mary; Krastoshevsky, Olga; Krause, Verena; Kumar, Ravinesh A.; Grozeva, Detelina; Malhotra, Dheeraj; Walsh, Tom (November 2009). "Microduplications of 16p11.2 are associated with schizophrenia". Nature Genetics. 41 (11): 1223–1227. doi:10.1038/ng.474. ISSN 1546-1718. PMC 2951180. PMID 19855392.
- Van, Lily; Boot, Erik; Bassett, Anne S. (May 2017). "Update on the 22q11.2 deletion syndrome and its relevance to schizophrenia". Current Opinion in Psychiatry. 30 (3): 191–196. doi:10.1097/YCO.0000000000000324. ISSN 1473-6578. PMID 28230630. S2CID 2026063.
External links
 Media related to Animal model of schizophrenia at Wikimedia Commons
Media related to Animal model of schizophrenia at Wikimedia Commons