Okamoto syndrome
Okamoto syndrome (OS), also known as Au–Kline syndrome (AKS), is a very rare autosomal dominant genetic condition characterised by congenital hydronephrosis, low muscle tone, heart defects, intellectual disability and characteristic facial features.[4][6] Those affected often have neurological and skeletal abnormalities, as well as frequent urinary tract infections. Language and walking are usually delayed. Facial features include prominent, downturned ears, an open, downturned mouth and drooping eyelids (ptosis).[4][5]
| Okamoto syndrome | |
|---|---|
| Other names | Au–Kline syndrome (AKS),[1] neurodevelopmental disorder–craniofacial dysmorphism–cardiac defect–skeletal anomalies syndrome,[2] congenital hydronephrosis with cleft palate, characteristic facies, hypotonia and mental retardation[3] |
 | |
| Boy with Okamoto syndrome, showing the characteristic facial features | |
| Specialty | Medical genetics |
| Symptoms | Congenital hydronephrosis, congenital heart defects, intellectual disability, dysautonomia, characteristic facial features[4] |
| Complications | Urinary tract infections[4] |
| Causes | Genetic (autosomal dominant mutation in HNRNPK)[4] |
| Diagnostic method | Based on symptoms, genetic testing[4] |
| Treatment | Symptomatic[4] |
| Prognosis | Not yet certain. Most patients have at least lived through childhood; mortality in infancy in a minority.[5] |
| Frequency | Not yet known. 26 individuals known to be affected as of May 2019.[1][4] |
The syndrome is caused by mutations in the HNRNPK gene, which codes for heterogeneous nuclear ribonucleoprotein K. This protein is involved in the process of DNA transcription and translation into proteins. A mutation in this gene impairs DNA transcription, disrupting some developmental processes.[4][7] As an autosomal dominant disorder, only one faulty copy of the gene is required for the condition to occur. The syndrome is typically diagnosed based on the physical symptoms and then confirmed by genetic testing.[4][5]
Treatment has centred around the symptoms. Sign language and assistive language technology can aid communication.[4][7] The prognosis is not yet fully known, due to the lack of patients in literature, however most of the patients have at least lived through childhood. The urinary system defects have been the most significant contributors to mortality.[5] As of May 2019, 26 individuals worldwide were known to be affected.[1][4] The syndrome was first described in 1997 by Nobuhiko Okamoto et al.,[8] and the gene responsible was first identified in 2015 by Ping-Yee Billie Au, Antonie D. Kline et al.[7] In 2019, Okamoto proposed that Au–Kline syndrome and Okamoto syndrome were synonymous.[1]
Signs and symptoms
Kidneys
Individuals with Okamoto syndrome are usually born with hydronephrosis, or dilation of the internal structures of the kidneys, due to narrowing (stenosis) of the passage between the kidneys and the ureters (the ureteropelvic junction), leading to a build-up of urine. There is also often vesicoureteral reflux, in which urine passes backwards from the bladder to the ureters, and frequent urinary tract infections.[3][4][5]
Heart
Individuals with Okamoto syndrome are typically born with heart defects, which can include aortic valve stenosis, atrial or ventricular septal defect, bicuspid aortic valve or patent ductus arteriosus.[3][4][7]
Physical features
The syndrome has a characteristic facial appearance which is similar to that of Kabuki syndrome, including prominent, downward-displaced ears that are underdeveloped, long eyelids, epicanthic folds, a short, broad nose, an open, downturned mouth and a deep groove in the midline of the tongue.[4] Cleft palate occurs in about half of those affected.[4] There is sometimes webbing of the neck or bulging eyes, and less commonly there is excessive hair on the forehead or other parts of the body or a unibrow.[3][8] Individuals with the syndrome may also have a broad first toe and crowding of the toes,[1][7][8] and at least two affected individuals have had polydactyly of the fifth digit (postaxial polydactyly).[4][7] Some of those affected have had undescended testicles (cryptorchidism).[1][4] A small minority of those affected have had congenital joint contractures such as club foot.[1]
Neurological
Those with Okamoto syndrome typically have severe mental disability and are usually born with microcephaly.[1][3][4] There are typically language and walking delays, and those affected have very low muscle tone and decreased reflexes.[3][4][8] They may have neural tube defects such as lipomyelomeningocele (a form of spina bifida) or may have syringomyelia (a cyst in the spinal cord).[3][4][7] Those with the syndrome may also have symptoms of dysautonomia (impairments in the autonomic nervous system), including gastrointestinal dysmotility, contributing to gastroesophageal reflux disease, or neurogenic bladder dysfunction, in which bladder control is limited.[3][4][5] Dysautonomia has also led to high pain tolerance and reduced sweating in some patients.[4][5] Some of those with the syndrome have been found to have an underdeveloped corpus callosum, the main band of white matter that connects the two cerebral hemispheres.[3][4][5]
Skeletal
Okamoto syndrome patients often have skeletal problems such as scoliosis or hip dysplasia, which can lead to hip dislocation.[1][3][4] They may be born with congenital vertebral anomalies; parts of the spine may be fused and fail to segment.[4] There may also be extra vertebrae in the lower back.[5][7] Some of those affected have been reported to have premature fusion of the skull bones (craniosynostosis), particularly those across the midline and at the front of the skull. This has led to an elongated skull shape known as scaphocephaly as well as a ridge on the forehead known as a metopic ridge.[4]
Growth
Those affected may be born with low weight and size[7][8] and may display stunted growth in childhood,[3] although this symptom has been variable and not in every individual with Okamoto syndrome.[4][5]
Hearing and eyesight
A minority of those with Okamoto syndrome have had hearing loss of both sensorineural and conductive types, and a smaller minority have had optic nerve abnormalities.[4][5][7]
Cause
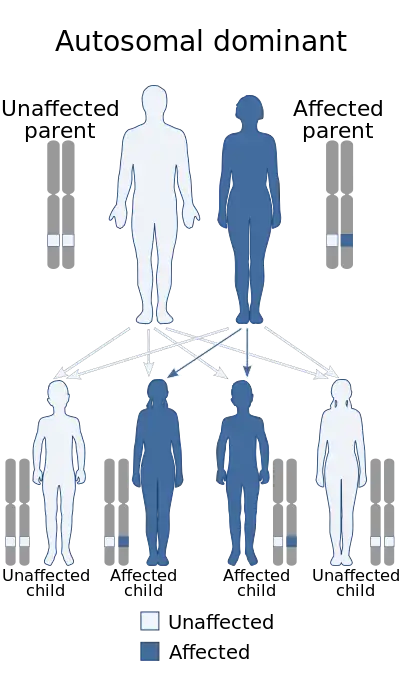
Okamoto syndrome is caused by a mutation in the HNRNPK gene, located on chromosome 9 at position q21.32.[1][4] HNRNPK codes for heterogeneous nuclear ribonucleoprotein K. Ribonucleoproteins are proteins that form complexes with RNA, and they are involved in the transcription of DNA into RNA in the nucleus. They usually bind with precursor messenger RNA (pre-mRNA), which is messenger RNA that has not yet had its introns removed during splicing to be ready for translation. A mutation in one of the two copies of HNRNPK results in defects in DNA transcription, and therefore some developmental processes are disrupted or not completed.[4][7]
Mutations in HNRNPK that have been reported include nonsense mutations, in which the protein is truncated and usually non-functional as a result, frameshift mutations, splice-site mutations and microdeletions encompassing the gene. Rarely, missense mutations, in which one amino acid in the protein is replaced by another, have also been reported.[4]
Deletions in the region encompassing HNRNPK have been found in the cells of acute myeloid leukemia in approximately 2% of cases.[9] Acute myeloid leukemia cells are immature white blood cells (myeloblasts) that remain in the stem-cell stage, dividing continually. Additionally, a 2015 study found that a majority of mice who had one of their HNRNPK genes artificially knocked out developed myeloid cancers, with a third developing lymphoid cancers and 4% developing hepatocellular carcinomas. The mice were also smaller, had less developed organs and had higher postnatal mortality (30%). The median lifespan of the mice that survived was less than 50% that of wild-type mice.[9] However, blood cancers had not yet been detected in any of the Okamoto syndrome patients as of 2018.[4][5]
Mutations in both copies of HNRNPK are embryonic lethal in mice. Mice with both copies of the gene knocked out die before the 14th day of embryonic development.[9]
Diagnosis
The condition has usually been diagnosed based on the physical symptoms and confirmed by genetic testing.[4] Methods have included whole exome sequencing and comparative genomic hybridization (for microdeletions). Sanger sequencing can confirm the nature of the mutation.[5]
Treatment
Treatment is focussed on the symptoms. Atrial or ventricular septal defects are usually treated with observation but can be surgically corrected in severe cases. Some patients have been able to use sign language or assistive language devices to facilitate communication.[4][7]
Prognosis
The prognosis of the disorder is not yet fully known. A minority of patients have died in infancy due to complications from their urinary system defects, including infections in Okamoto's first two patients,[1] however most have lived through childhood and into adolescence.[5] Motor and language skills typically improve as the patient ages. The prognosis in adulthood is not yet known, due to the lack of known patients in this age group.[5]
As an autosomal dominant condition, there is little risk of recurrence in future conceptions from unaffected parents. However, there is a slight possibility (around 1%) due to germline mosaicism, a phenomenon in which some sperm cell precursors have the mutation and others don't. Genetic counselling may be offered for this.[4]
Epidemiology
The prevalence of the disorder is as yet unknown. As of May 2019, 26 individuals worldwide were known to be affected, with 13 of these reported in literature, mostly from 2010 to 2019.[1][4]
History
Okamoto syndrome was first described in 1997 by Nobuhiko Okamoto et al. from the Department of Medical Genetics at Osaka Women's and Children's Hospital after observing very similar symptoms and physical features in two unrelated Japanese infants. Both infants had congenital hydronephrosis due to ureteropelvic junction stenosis, low muscle tone, developmental delay and characteristic facial features including an open mouth and low-set ears.[8]
Au–Kline syndrome was first described in 2015 by Ping-Yee Billie Au, Antonie D. Kline et al. after mutations in HNRNPK were found in two individuals with similar symptoms at their respective practices in Calgary, Alberta, Canada and Baltimore, Maryland, United States. The practices were united with each other after both submitted the gene as a candidate to the online service GeneMatcher, which matched them together and allowed them to confirm the syndrome.[7]
In 2019, Okamoto proposed that Au–Kline syndrome and Okamoto syndrome were synonymous, because a mutation in the HNRNPK gene had been found in a new Okamoto syndrome patient, and the symptoms were virtually identical.[1]
See also
References
- Okamoto, Nobuhiko (May 2019). "Okamoto syndrome has features overlapping with Au-Kline syndrome and is caused by HNRNPK mutation". American Journal of Medical Genetics. Part A. 179 (5): 822–826. doi:10.1002/ajmg.a.61079. ISSN 1552-4833. PMID 30793470. S2CID 73496854.
- "Orphanet: Neurodevelopmental disorder craniofacial dysmorphism cardiac defect skeletal anomalies syndrome". www.orpha.net. Retrieved 9 December 2019.
- "Okamoto syndrome | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Retrieved 2 December 2019.
- Au, Ping-Yee Billie; Innes, A. Micheil; Kline, Antonie D. (2019), Adam, Margaret P.; Ardinger, Holly H.; Pagon, Roberta A.; Wallace, Stephanie E. (eds.), "Au-Kline Syndrome", GeneReviews®, University of Washington, Seattle, PMID 30998304, retrieved 2 December 2019
- Au, P. Y. Billie; Goedhart, Caitlin; Ferguson, Marcia; Breckpot, Jeroen; Devriendt, Koenraad; Wierenga, Klaas; Fanning, Elizabeth; Grange, Dorothy K.; Graham, Gail E.; Galarreta, Carolina; Jones, Marilyn C. (September 2018). "Phenotypic spectrum of Au-Kline syndrome: a report of six new cases and review of the literature". European Journal of Human Genetics. 26 (9): 1272–1281. doi:10.1038/s41431-018-0187-2. ISSN 1476-5438. PMC 6117294. PMID 29904177.
- "Orphanet: Okamoto syndrome". www.orpha.net. Retrieved 30 November 2019.
- Au, P.Y. Billie; You, Jing; Caluseriu, Oana; Schwartzentruber, Jeremy; Majewski, Jacek; Bernier, Francois P.; Ferguson, Marcia; Valle, David; Parboosingh, Jillian S.; Sobreira, Nara; Innes, A. Micheil (October 2015). "GeneMatcher Aids in the Identification of a New Malformation Syndrome with Intellectual Disability, Unique Facial Dysmorphisms, and Skeletal and Connective Tissue Abnormalities Caused by De Novo Variants in HNRNPK". Human Mutation. 36 (10): 1009–1014. doi:10.1002/humu.22837. ISSN 1059-7794. PMC 4589226. PMID 26173930.
- Okamoto, Nobuhiko; Matsumoto, Fumi; Shimada, Kenji; Satomura, Kenichi (1997). "New MCA/MR syndrome with generalized hypotonia, congenital hydronephrosis, and characteristic face". American Journal of Medical Genetics. 68 (3): 347–349. doi:10.1002/(SICI)1096-8628(19970131)68:3<347::AID-AJMG18>3.0.CO;2-T. ISSN 1096-8628. PMID 9024570.
- Gallardo, Miguel; Lee, Hun Ju; Zhang, Xiaorui; Bueso-Ramos, Carlos; Pageon, Laura R.; McArthur, Mark; Multani, Asha; Nazha, Aziz; Manshouri, Taghi; Parker-Thornburg, Jan; Rapado, Inmaculada (12 October 2015). "hnRNP K Is a Haploinsufficient Tumor Suppressor that Regulates Proliferation and Differentiation Programs in Hematologic Malignancies". Cancer Cell. 28 (4): 486–499. doi:10.1016/j.ccell.2015.09.001. ISSN 1878-3686. PMC 4652598. PMID 26412324.
External links
- Au–Kline syndrome at the US National Institutes of Health (NIH) Genetics Home Reference