Zellweger syndrome
Zellweger syndrome is a rare congenital disorder characterized by the reduction or absence of functional peroxisomes in the cells of an individual.[1] It is one of a family of disorders called Zellweger spectrum disorders which are leukodystrophies. Zellweger syndrome is named after Hans Zellweger (1909–1990), a Swiss-American pediatrician, a professor of pediatrics and genetics at the University of Iowa who researched this disorder.[2][3]
| Zellweger syndrome | |
|---|---|
| Other names | Cerebrohepatorenal syndrome |
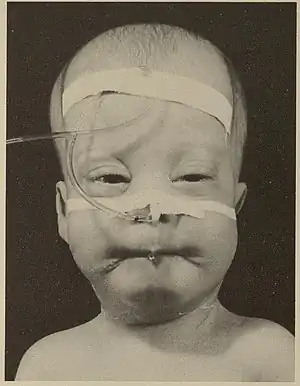 | |
| Infant with Zellweger syndrome | |
| Specialty | Medical genetics |
| Complications | pneumonia and respiratory distress. |
Signs and symptoms
Zellweger syndrome is one of three peroxisome biogenesis disorders which belong to the Zellweger spectrum of peroxisome biogenesis disorders (PBD-ZSD).[4] The other two disorders are neonatal adrenoleukodystrophy (NALD), and infantile Refsum disease (IRD).[5][6] Although all have a similar molecular basis for disease, Zellweger syndrome is the most severe of these three disorders.[7]
Zellweger syndrome is associated with impaired neuronal migration, neuronal positioning, and brain development.[4] In addition, individuals with Zellweger syndrome can show a reduction in central nervous system (CNS) myelin (particularly cerebral), which is referred to as hypomyelination. Myelin is critical for normal CNS functions, and in this regard, serves to insulate nerve fibers in the brain. Patients can also show postdevelopmental sensorineuronal degeneration that leads to a progressive loss of hearing and vision.[4]
Zellweger syndrome can also affect the function of many other organ systems. Patients can show craniofacial abnormalities (such as a high forehead, hypoplastic supraorbital ridges, epicanthal folds, midface hypoplasia, and a large fontanel), hepatomegaly (enlarged liver), chondrodysplasia punctata (punctate calcification of the cartilage in specific regions of the body), eye abnormalities, and renal cysts.[4] Newborns may present with profound hypotonia (low muscle tone), seizures, apnea, and an inability to eat.[4][7]
Cause

Zellweger syndrome is an autosomal recessive disorder caused by mutations in genes that encode peroxins, proteins required for the normal assembly of peroxisomes. Most commonly, patients have mutations in the PEX1, PEX2, PEX3, PEX5, PEX6, PEX10, PEX12, PEX13, PEX14, PEX16, PEX19, or PEX26 genes.[8] In almost all cases, patients have mutations that inactivate or greatly reduce the activity of both the maternal and paternal copies of one these aforementioned PEX genes.As a result of impaired peroxisome function, an individual's tissues and cells can accumulate very long chain fatty acids (VLCFA) and branched chain fatty acids (BCFA) that are normally degraded in peroxisomes. The accumulation of these lipids can impair the normal function of multiple organ systems, as discussed above. In addition, these individuals can show deficient levels of plasmalogens, ether-phospholipids that are especially important for brain and lung function.
Diagnosis
In addition to genetic tests involving the sequencing of PEX genes,[9][10] biochemical tests have proven highly effective for the diagnosis of Zellweger syndrome and other peroxisomal disorders. Typically, Zellweger syndrome patients show elevated very long chain fatty acids in their blood plasma. Cultured primary skin fibroblasts obtained from patients show elevated very long chain fatty acids, impaired very long chain fatty acid beta-oxidation, phytanic acid alpha-oxidation, pristanic acid alpha-oxidation, and plasmalogen biosynthesis.[4]
Treatment
The nutrient malabsorption resulting from a lack of bile acids has resulted in elemental formula being suggested for feeding. They are low in fat, with less than 3 per cent of calories being derived from long-chain triglycerides (LCT). However, reducing dietary very long chain fatty acids (VLCFA) has not been shown to reduce blood VLCFA levels,[11][12] likely because humans can endogenously produce most VLCFA. Plasma VLCFA levels are decreased when dietary VLCFA is reduced in conjunction with supplementation of Lorenzo's oil (a 4:1 mixture of glyceryl trioleate and glyceryl trierucate) in X-ALD patients.[13] Since docosahexaenoic acid (DHA) synthesis is impaired [14] [59], DHA supplementation was recommended, but a placebo-controlled study has since shown no clinical efficacy.[15] Due to defective bile acid synthesis, fat-soluble supplements of vitamins A, D, E, and K are recommended.
Prognosis
Currently, no cure for Zellweger syndrome is known, nor is there a standard course of treatment. Infections should be guarded against to prevent such complications as pneumonia and respiratory distress. Other treatment is symptomatic and supportive. Patients usually do not survive beyond one year of age.[4]
References
- Brul, S.; Westerveld, A.; Strijland, A.; Wanders, R.; Schram, A.; Heymans, H.; Schutgens, R.; Van Den Bosch, H.; Tager, J. (June 1988). "Genetic heterogeneity in the cerebrohepatorenal (Zellweger) syndrome and other inherited disorders with a generalized impairment of peroxisomal functions. A study using complementation analysis". Journal of Clinical Investigation (Free full text). 81 (6): 1710–1715. doi:10.1172/JCI113510. PMC 442615. PMID 2454948.
- Zellweger's syndrome at Who Named It?
- Wiedemann, H. R. (1991). "Hans-Ulrich Zellweger (1909-1990)". European Journal of Pediatrics. 150 (7): 451. doi:10.1007/BF01958418. PMID 1915492. S2CID 34905299.
- Steinberg, S.; Dodt, G.; Raymond, G.; Braverman, N.; Moser, A.; Moser, H. (2006). "Peroxisome biogenesis disorders". Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 1763 (12): 1733–48. doi:10.1016/j.bbamcr.2006.09.010. PMID 17055079.
- GeneReviews: Peroxisome Biogenesis Disorders, Zellweger Syndrome Spectrum
- Krause, C.; Rosewich, H.; Thanos, M.; Gärtner, J. (2006). "Identification of novel mutations in PEX2, PEX6, PEX10, PEX12, and PEX13 in Zellweger spectrum patients". Human Mutation. 27 (11): 1157. doi:10.1002/humu.9462. PMID 17041890. S2CID 9905589.
- Raymond, G. V.; Watkins, P.; Steinberg, S.; Powers, J. (2009). "Peroxisomal Disorders". Handbook of Neurochemistry and Molecular Neurobiology. pp. 631–670. doi:10.1007/978-0-387-30378-9_26. ISBN 978-0-387-30345-1.
- Online Mendelian Inheritance in Man (OMIM): Zellweger syndrome; ZS - 214100
- Steinberg, S.; Chen, L.; Wei, L.; Moser, A.; Moser, H.; Cutting, G.; Braverman, N. (2004). "The PEX Gene Screen: molecular diagnosis of peroxisome biogenesis disorders in the Zellweger syndrome spectrum". Molecular Genetics and Metabolism. 83 (3): 252–263. doi:10.1016/j.ymgme.2004.08.008. PMID 15542397.
- Yik, W. Y.; Steinberg, S. J.; Moser, A. B.; Moser, H. W.; Hacia, J. G. (2009). "Identification of novel mutations and sequence variation in the Zellweger syndrome spectrum of peroxisome biogenesis disorders". Human Mutation. 30 (3): E467–E480. doi:10.1002/humu.20932. PMC 2649967. PMID 19105186.
- Van Duyn, MA; Moser, AE; Brown FR, 3rd; et al. (August 1984). "The design of a diet restricted in saturated very long-chain fatty acids: therapeutic application in adrenoleukodystrophy". The American Journal of Clinical Nutrition. 40 (2): 277–84. doi:10.1093/ajcn/40.2.277. PMID 6465061.
- Brown FR, 3rd; Van Duyn, MA; Moser, AB; et al. (October 1982). "Adrenoleukodystrophy: effects of dietary restriction of very long chain fatty acids and of administration of carnitine and clofibrate on clinical status and plasma fatty acids". The Johns Hopkins Medical Journal. 151 (4): 164–72. PMID 7120720.
- Moser, AB; Borel, J; Odone, A; et al. (March 1987). "A new dietary therapy for adrenoleukodystrophy: biochemical and preliminary clinical results in 36 patients". Annals of Neurology. 21 (3): 240–9. doi:10.1002/ana.410210305. PMID 2440378. S2CID 29043456.
- Martinez, M (26 June 1992). "Abnormal profiles of polyunsaturated fatty acids in the brain, liver, kidney and retina of patients with peroxisomal disorders". Brain Research. 583 (1–2): 171–82. doi:10.1016/s0006-8993(10)80021-6. PMID 1504825. S2CID 20508763.
- Paker, AM; Sunness, JS; Brereton, NH; et al. (31 August 2010). "Docosahexaenoic acid therapy in peroxisomal diseases: results of a double-blind, randomized trial". Neurology. 75 (9): 826–30. doi:10.1212/WNL.0b013e3181f07061. PMC 3013498. PMID 20805528.
External links
- Zellweger-Syndrome at NINDS
- Zellweger syndrome at NIH's Office of Rare Diseases
- Steinberg SJ, Raymond GV, Braverman NE, et al. (2020). Adam MP, Ardinger HH, Pagon RA, et al. (eds.). "Zellweger Spectrum Disorder". GeneReviews® [Internet]. University of Washington. PMID 20301621. NBK1448.