Contig
A contig (from contiguous) is a set of overlapping DNA segments that together represent a consensus region of DNA.[1] In bottom-up sequencing projects, a contig refers to overlapping sequence data (reads);[2] in top-down sequencing projects, contig refers to the overlapping clones that form a physical map of the genome that is used to guide sequencing and assembly.[3] Contigs can thus refer both to overlapping DNA sequences and to overlapping physical segments (fragments) contained in clones depending on the context.
Original definition of contig
In 1980, Staden [4] wrote: In order to make it easier to talk about our data gained by the shotgun method of sequencing we have invented the word "contig". A contig is a set of gel readings that are related to one another by overlap of their sequences. All gel readings belong to one and only one contig, and each contig contains at least one gel reading. The gel readings in a contig can be summed to form a contiguous consensus sequence and the length of this sequence is the length of the contig.
Sequence contigs
A sequence contig is a continuous (not contiguous) sequence resulting from the reassembly of the small DNA fragments generated by bottom-up sequencing strategies. This meaning of contig is consistent with the original definition by Rodger Staden (1979).[5] The bottom-up DNA sequencing strategy involves shearing genomic DNA into many small fragments ("bottom"), sequencing these fragments, reassembling them back into contigs and eventually the entire genome ("up"). Because current technology allows for the direct sequencing of only relatively short DNA fragments (300–1000 nucleotides), genomic DNA must be fragmented into small pieces prior to sequencing.[6] In bottom-up sequencing projects, amplified DNA is sheared randomly into fragments appropriately sized for sequencing. The subsequent sequence reads, which are the data that contain the sequences of the small fragments, are put into a database. The assembly software[6] then searches this database for pairs of overlapping reads. Assembling the reads from such a pair (including, of course, only one copy of the identical sequence) produces a longer contiguous read (contig) of sequenced DNA. By repeating this process many times, at first with the initial short pairs of reads but then using increasingly longer pairs that are the result of previous assembly, the DNA sequence of an entire chromosome can be determined.
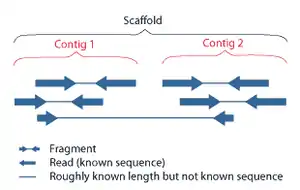
Today, it is common to use paired-end sequencing technology where both ends of consistently sized longer DNA fragments are sequenced. Here, a contig still refers to any contiguous stretch of sequence data created by read overlap. Because the fragments are of known length, the distance between the two end reads from each fragment is known.[7] This gives additional information about the orientation of contigs constructed from these reads and allows for their assembly into scaffolds in a process called scaffolding.
Scaffolds consist of overlapping contigs separated by gaps of known length. The new constraints placed on the orientation of the contigs allows for the placement of highly repeated sequences in the genome. If one end read has a repetitive sequence, as long as its mate pair is located within a contig, its placement is known.[7] The remaining gaps between the contigs in the scaffolds can then be sequenced by a variety of methods, including PCR amplification followed by sequencing (for smaller gaps) and BAC cloning methods followed by sequencing for larger gaps.[2]
BAC contigs
Contig can also refer to the overlapping clones that form a physical map of a chromosome when the top-down or hierarchical sequencing strategy is used.[1] In this sequencing method, a low-resolution map is made prior to sequencing in order to provide a framework to guide the later assembly of the sequence reads of the genome. This map identifies the relative positions and overlap of the clones used for sequencing. Sets of overlapping clones that form a contiguous stretch of DNA are called contigs; the minimum number of clones that form a contig that covers the entire chromosome comprise the tiling path that is used for sequencing. Once a tiling path has been selected, its component BACs are sheared into smaller fragments and sequenced. Contigs therefore provide the framework for hierarchical sequencing.[3]
The assembly of a contig map involves several steps. First, DNA is sheared into larger (50–200kb) pieces, which are cloned into BACs or PACs to form a BAC library. Since these clones should cover the entire genome/chromosome, it is theoretically possible to assemble a contig of BACs that covers the entire chromosome.[1] Reality, however, is not always ideal. Gaps often remain, and a scaffold—consisting of contigs and gaps—that covers the map region is often the first result.[1] The gaps between contigs can be closed by various methods outlined below.
Construction of BAC contigs
BAC contigs are constructed by aligning BAC regions of known overlap via a variety of methods. One common strategy is to use sequence-tagged site (STS) content mapping to detect unique DNA sites in common between BACs. The degree of overlap is roughly estimated by the number of STS markers in common between two clones, with more markers in common signifying a greater overlap.[2] Because this strategy provides only a very rough estimate of overlap, restriction digest fragment analysis, which provides a more precise measurement of clone overlap, is often used.[2] In this strategy, clones are treated with one or two restriction enzymes and the resulting fragments separated by gel electrophoresis. If two clones, they will likely have restriction sites in common, and will thus share several fragments.[3] Because the number of fragments in common and the length of these fragments is known (the length is judged by comparison to a size standard), the degree of overlap can be deduced to a high degree of precision.
Gaps between contigs
Gaps often remain after initial BAC contig construction. These gaps occur if the Bacterial Artificial Chromosome (BAC) library screened has low complexity, meaning it does not contain a high number of STS or restriction sites, or if certain regions were less stable in cloning hosts and thus underrepresented in the library.[1] If gaps between contigs remain after STS landmark mapping and restriction fingerprinting have been performed, the sequencing of contig ends can be used to close these gaps. This end-sequencing strategy essentially creates a novel STS with which to screen the other contigs. Alternatively, the end sequence of a contig can be used as a primer to primer walk across the gap.[2]
See also
References
- Gregory, S. Contig Assembly. Encyclopedia of Life Sciences, 2005.
- Gibson, Greg; Muse, Spencer V. (2009). A Primer of Genome Science (3rd ed.). Sinauer Associates. p. 84. ISBN 978-0-878-93236-8.
- Dear, P. H. Genome Mapping. Encyclopedia of Life Sciences, 2005. doi:10.1038/npg.els.0005353.
- Staden, R (1980). "A new computer method for the storage and manipulation of DNA gel reading data". Nucleic Acids Research. 8 (16): 3673–3694. doi:10.1093/nar/8.16.3673. PMC 324183. PMID 7433103.
- Staden R (1979). "A strategy of DNA sequencing employing computer programs". Nucleic Acids Research. 6 (7): 2601–2610. doi:10.1093/nar/6.7.2601. PMC 327874. PMID 461197.
- Dunham, I. Genome Sequencing. Encyclopedia of Life Sciences, 2005.
- Fullwood MJ, Wei C, Liu ET, et al. (2009). "Next-generation DNA sequencing of paired-end tags (PET) for transcriptome and genome analyses". Genome Research. 19 (4): 521–532. doi:10.1101/gr.074906.107. PMC 3807531. PMID 19339662.