Danishefsky Taxol total synthesis
The Danishefsky Taxol total synthesis in organic chemistry is an important third Taxol synthesis published by the group of Samuel Danishefsky in 1996[1] two years after the first two efforts described in the Holton Taxol total synthesis and the Nicolaou Taxol total synthesis. Combined they provide a good insight in the application of organic chemistry in total synthesis.
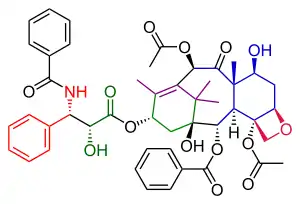
Danishefsky's route to Taxol has many similarities with that of Nicolaou. Both are examples of convergent synthesis with a coupling of the A and the C ring from two precursors. The main characteristic of the Danishefsky variant is the completion of the oxetane D ring onto the cyclohexanol C ring prior to the construction of the 8-membered B ring. The most prominent starting material is the (+) enantiomer of the Wieland-Miescher ketone. This compound is commercially available as a single enantiomer and the single chiral group present in this molecule is able to drive the entire sequence of organic reactions to a single optically active Taxol endproduct. The final step, the tail addition is identical to that of Nicolaou and is based on Ojima chemistry.
In terms of raw material shopping, this taxol molecule consists of the aforementioned Wieland-Miescher ketone, 2-methyl-3-pentanone, lithium aluminium hydride, osmium tetroxide, phenyllithium, pyridinium chlorochromate, the Corey-Chaykovsky reagent and acryloyl chloride. Key chemical transformations are the Johnson-Corey-Chaykovsky reaction and the Heck reaction.
Retrosynthesis
Taxol resulted from the tail addition of the Ojima lactam to alcohol 51, which is baccatin III (the original target molecule of the Danishefsky synthesis). Alcohol 51 was derived from the allylic oxidation of α-acylketone 49. Compound 49 was ultimately derived from the Heck reaction of enol triflate 38, which was used to close the B-ring. Enol triflate 38 resulted from a rearrangement of compound 31 after protection of its hydroxyl group. Compound 31 was derived from the connection of the A and C rings with aldehyde 21 combining with the vinyllithium reagent derived from cyanohydrin 29. Cyanohydrin 29 originated as the ethyl isopropyl ketone (22). Aldehyde 21 was obtained from compound 17, which was the product of the opening of ketal 12. Ketal 12 was ultimately derived from the Wieland-Miescher ketone (1).
 Ring D synthesis scheme 1 |
| Retrosynthesis |
|---|
D Ring synthesis
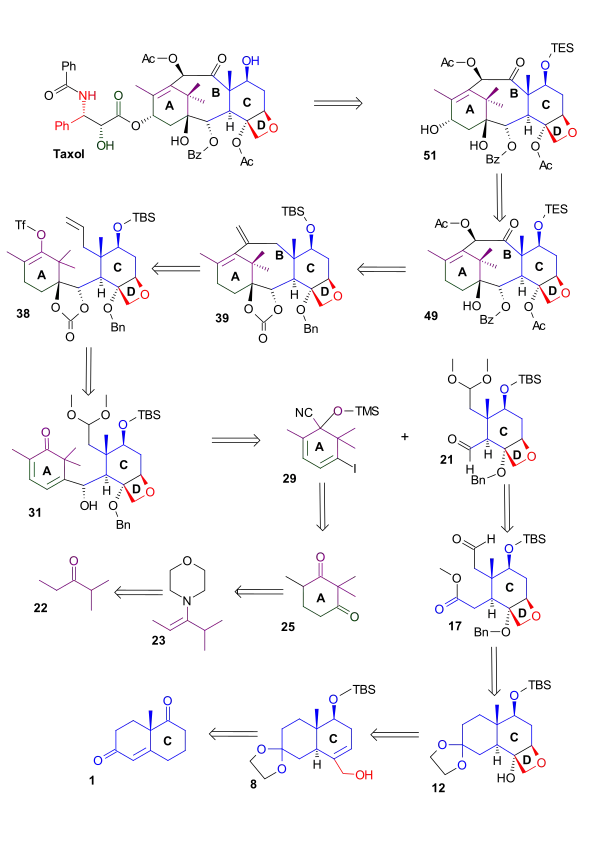
Scheme 1 shows the synthesis of the oxetane D ring from the C ring starting from the (+) enantiomer of the Wieland-Miescher ketone (1). Reduction of this diketone with sodium borohydride provided unsaturated ketoalcohol 2, which was protected as an acetate. Formation of the ketal was accompanied by alkene rearrangement. The acetyl group was replaced by a tert-butyldimethylsilyl protecting group. Hydroboration followed by oxidation with hydrogen peroxide gave alcohol 5. The hydroxyl group was then oxidized to a carbonyl group giving ketone 6 by action of pyridinium dichromate. With all the sensitive functional groups protected, the methylene group required for the oxetane ring D was then provided by the Corey-Chaykovsky reagent, which converted the carbonyl group to an epoxide (7). Treatment of this epoxide with aluminium isopropoxide gave allylic alcohol 8. Two more hydroxyl groups were added by oxidation of the newly formed double bond with a catalytic amount of osmium tetroxide in the presence of N-methylmorpholine N-oxide. This reaction lacked stereospecificity and the yield of triol 9 with the correct stereochemistry was therefore reduced. The primary alcohol was protected as a silyl ether and the secondary alcohol was activated as a triflate (11). Heating this trimethylsilyl protected triflate in refluxing ethlyene glycol closed the ring to give oxetane 12.
 Ring D synthesis scheme 1 |
| Scheme 1 |
|---|
Preparation for AB ring synthesis
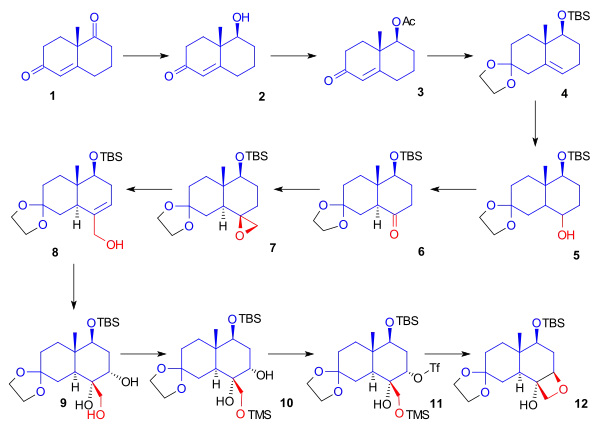
In the next phase (Scheme 2), starting from ketal 12, the cyclohexane ring was cleaved to provide two anchoring points for fusion with the A ring. Alcohol 12 was protected by a benzyl group. The acetonide protecting group was removed from the ketone. Ketone 14 was converted to silyl enol ether 15 by reaction with trimethylsilyl triflate, and a modified Rubottom oxidation using 3,3-dimethyldioxirane followed by a treatment with camphorsulfonic acid introduced a hydroxyl group alpha to the ketone. Ring opening by oxidative cleavage with lead tetraacetate in methanol gave compound 17. In the next step, the aldehyde was protected as a dimethyl acetal, and the ester was reduced to give primary alcohol 18. The hydroxyl group was converted in a Grieco elimination to the selenide (19), which on oxidation with hydrogen peroxide gave alkene 20. Ozonolysis with ozone and triphenylphosphine provided aldehyde 21.
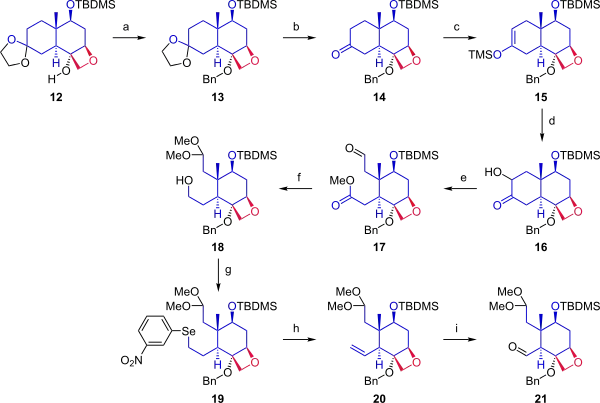 Scheme 2 Danishefsky Taxol total synthesis C ring |
| Scheme 2 |
|---|
AB ring synthesis
For this synthesis (Scheme 3) the morpholine enamine of ethyl isopropyl ketone was reacted with acryloyl chloride in a combined nucleophilic conjugate addition and nucleophilic acyl substitution to give after hydrolysis diketone 25. Reaction with hydrazine in triethylamine and ethanol afforded hydrazone 26. After an unusual hydrazone iodination that also involved iodination alpha to a carbonyl group and elimination of HI, fully conjugated vinyl iodide 28 was produced in an unexpected dehydrogenation. The ketone was converted into cyanohydrin 29 with trimethylsilyl cyanide, potassium cyanide and a crown ether.
|
|
| Scheme 3 |
|---|
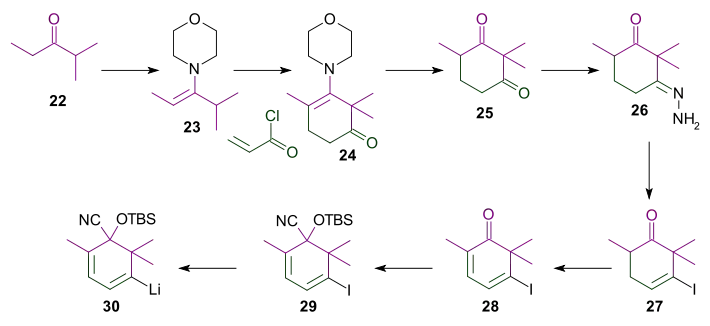
As shown in Scheme 4, the bottom part of the taxol B ring synthesis involved the reaction of ring C aldehyde group of 21. The ketone group was deprotected by action of tetra-n-butylammonium fluoride, and the double bond was epoxidized with meta-chloroperoxybenzoic acid to epoxide 32. This epoxide was then hydrogenated with hydrogen over palladium on carbon to give diol 33, which was protected in the next step as the cyclic carbonate ester (34).
L-Selectride reduction of enone 34 gave ketone 35. The ketone was converted into vinyl triflate 36 using phenyl triflimide and potassium hexamethyldisilazide in tetrahydrofuran at −78 °C. This vinyl triflate was one of the functional groups required for the Heck reaction. For the generation of the other reactive group the acetal group was removed to give aldehyde 37 which was subsequently converted to the terminal alkene 38 in a Wittig reaction involving methylenetriphenylphosphorane. The intramolecular Heck reaction involved tetrakis(triphenylphosphine)palladium(0) and potassium carbonate in acetonitrile at reflux to give diene 39 and to complete the formation of the B ring.
|
|
| Scheme 4 |
|---|
B ring elaboration
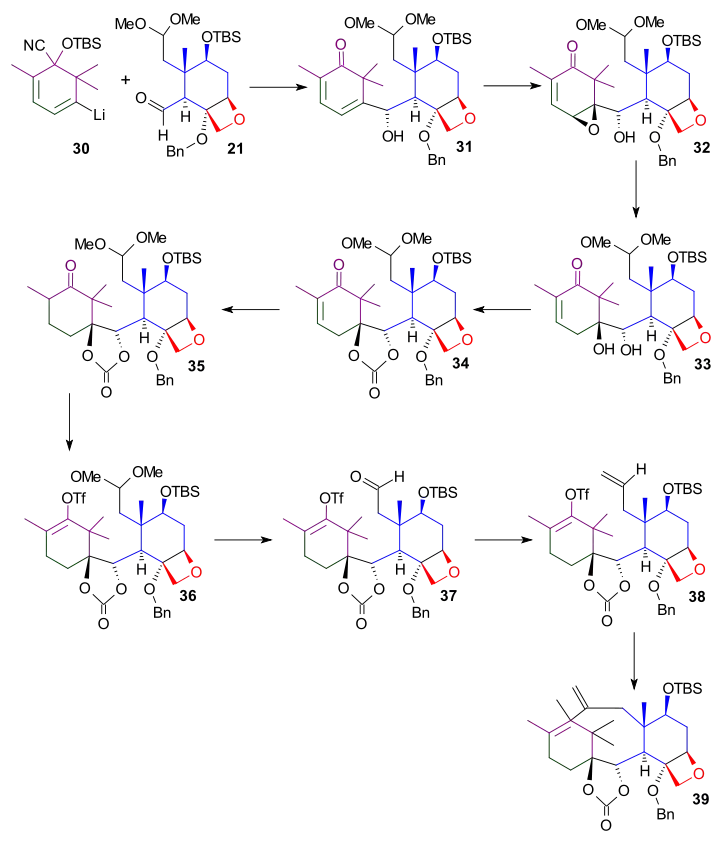
The second part of the B ring synthesis (Scheme 5) was concerned with correct chemistry for the newly formed ethylene bridge connecting the A and C rings. After Scheme 4, this bridge contained an exocyclic methylene group, but in the ultimate taxol molecule this bridge is an α-acylketone. The required conversion was accomplished in the next 10 steps.
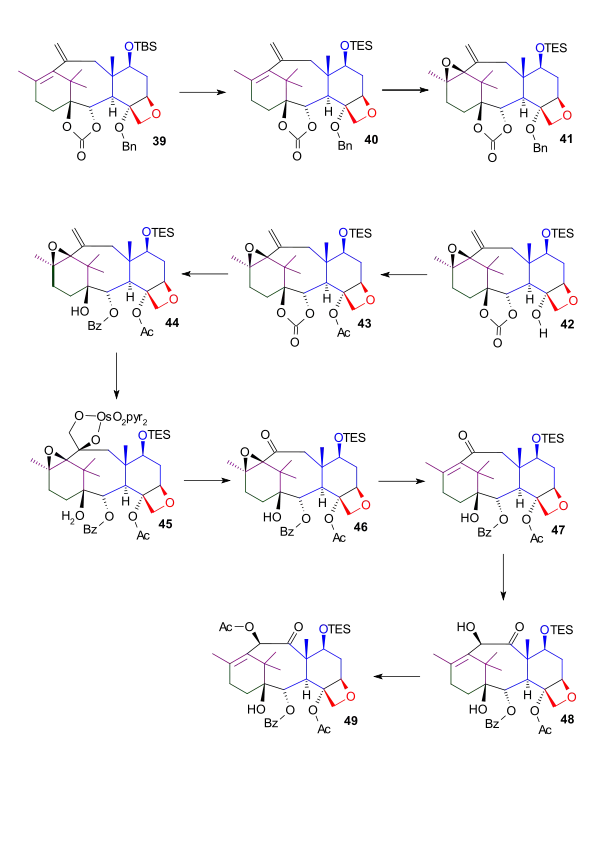
The tert-butylsilyl protecting group in diene 39 was not compatible in later reactions and was replaced by a triethylsilyl. Epoxidation of diene 40 with meta-chloroperoxybenzoic acid gave the oxirane ring. This served solely as a protecting group in preparation for modifications of the exocyclic alkene. In the next two steps, the benzyl protecting group in compound 41 was replaced by an acetyl group. Carbonate ester 43 was opened by reaction with phenyllithium to give alcohol 44. The cleavage of the exocyclic double bond was difficult and accomplished only with forcing conditions (19 equivalents of osmium tetroxide, 105 °C, 24 hours) by the putative osmate ester (45). Subsequent oxidative cleavage with lead tetraacetate gave ketone 46. The epoxide protecting group was removed with samarium (II) iodide[2] to give α-ß-unsaturated ketone 47. The enolate was formed by the reaction of ketone 47 with potassium tert-butoxide, and subsequent reaction with phenylseleninic anhydride followed by acylation gave α-acylketone 49.
 scheme 5 Danishefsky Taxol total synthesis B ring Part B |
| Scheme 5 |
|---|
Tail addition
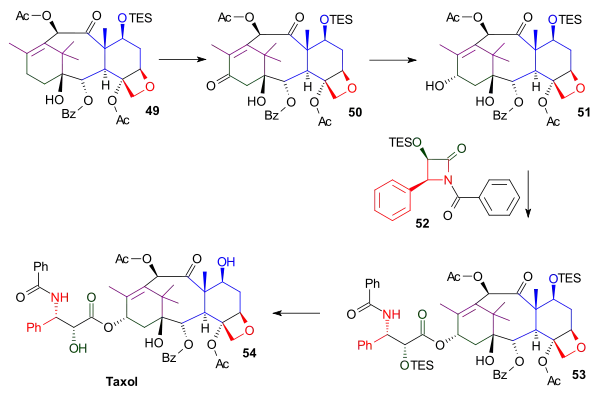
The tail addition step in this synthesis (Scheme 6) was identical to that in the Nicolaou tail addition and was based on Oijma chemistry. The A ring was functionalized with a hydroxyl group through pyridinium chlorochromate oxidation of α-acylketone 49 to form ketone 50. Subsequent reduction using sodium borohydride produced alcohol 51 . Reaction of this alcohol with the Ojima lactam 52 and a concluding silyl deprotection step at two triethyl silyl positions in compound 53 gave Taxol.
 Scheme 6 Danishefsky Taxol total synthesis Tail Addition |
| Scheme 6 |
|---|
Protecting groups
The synthesis makes use of various protecting groups as follows:
| Protecting group | Protection reagents | Deprotection reagents | Use in synthesis |
|---|---|---|---|
| Ac (acetate) | acetic anhydride, 4-(dimethylamino)pyridine and pyridine. | sodium ethoxide, ethanol | The hydroxyl group in alcohol 3 (Scheme 1) was protected as an acetate during the subsequent alkene rearrangement. The acetate was removed by a tert-butyldimethylsilyl protecting group (ketone 6). |
| Acetonide | ethylene glycol, naphthalenesulfonic acid | p-toluenesulfonic acid, acetone, and water. | Ketone 3 (Scheme 1) was protected using an acetonide group, which was removed after the closure of ring D (ketone 14, Scheme 2). |
| Bn (benzyl) | benzyl chloride, pyridine | acetic anhydride, 4-(dimethylamino)pyridine, and pyridine | The hydroxyl group in ketal 12 (Scheme 2) was protected as a benzyl ether, which was replaced much later in the synthesis (alcohol 42, Scheme 5) |
| Carbonate ester (cyclic) | carbonyl diimidazole, sodium hydride, dimethylformamide | phenyllithium | Diol 33 was protected as a cyclic carbonate ester (Scheme 4). Treatment of the carbonate ester with phenyllithium deprotected one hydroxyl group and left a benzoyl group needed for Taxol on the other oxygen (alcohol 44, Scheme 5). |
| Dimethyl acetal | methanol, collidine p-toluenesulfonate | pyridinium tosylate | The carbonyl group in aldehyde 17 of Scheme 2 was protected as the dimethyl acetal in order to allow the addition of the A ring to the C ring (Scheme 4). The aldehyde is later deprotected using pyridinium tosylate later on in Scheme 4. |
| Epoxide | meta-chloroperoxybenzoic acid | samarium(II) iodide, acetic anhydride, tetrahydrofuran | One of the double-bonds of diene 39 (Scheme 5) was protected as an epoxide in preparation for modifications of the other alkene. The epoxide was present for 4 steps before being removed with samarium(II) oxide. |
| TBS (tert-butyldimethylsilyl) | tert-butyldimethylsilyl chloride, lutidine | tetra-n-butylammonium fluoride, tetrahydrofuran | The acetate protecting group required for the acetylation of enone 3 (Scheme 1) was replaced by the more robust tert-butyldimethylsilyl protecting group. This silyl group was removed much later, in order to have a more easily removed protecting group present for the final steps of the Taxol synthesis (compound 40, Scheme 5). |
| TES (triethylsilyl) [1] | triethylsilyl triflate, triethylamine, and dichloromethane. | hydrogen fluoride, pyridine, and acetonitrile | A triethylsilyl group replaced a tert-butyl silyl protecting group late in the synthesis (compound 40, Scheme 5). The TES group is later removed in the concluding silyl deprotection step that gives the final Taxol compound. |
| TES (triethylsilyl) [2] | see Ojima lactam | hydrogen fluoride, pyridine, and acetonitrile | The TES protecting group that was present in the Ojima lactam is removed in the concluding silyl deprotection step of the Taxol total synthesis. |
| TMS (trimethylsilyl) | trimethylsilyl cyanide, potassium cyanide, and a crown ether | tetra-n-butylammonium fluoride | The ketone of diene 28 (Scheme 3) was protected by the trimethylsilyl group, and was removed when the vinyl lithium 30 group of A ring was added to the C ring aldehyde group in 21 (Scheme 4). |
See also
External links
References
- Samuel J. Danishefsky; John J. Masters; Wendy B. Young; J. T. Link; Lawrence B. Snyder; Thomas V. Magee; David K. Jung; Richard C. A. Isaacs; William G. Bornmann; Cheryl A. Alaimo; Craig A. Coburn; Martin J. Di Grandi (1996). "Total Synthesis of Baccatin III and Taxol". J. Am. Chem. Soc. 118 (12): 2843–2859. doi:10.1021/ja952692a.
- Molander, G. "Application of lanthanide reagents in organic synthesis" Chem. Rev. 1992, 92 (1), 29–68 doi:10.1021/cr00009a002