Epigenetics of anxiety and stress–related disorders
Epigenetics of anxiety and stress–related disorders is the field studying the relationship between epigenetic modifications of genes and anxiety and stress-related disorders, including mental health disorders such as generalized anxiety disorder (GAD), post-traumatic stress disorder, obsessive-compulsive disorder (OCD), and more. These change can lead to transgenerational stress inheritance.[1]
Epigenetic modifications play a role in the development and heritability of these disorders and related symptoms. For example, regulation of the hypothalamus-pituitary-adrenal axis by glucocorticoids plays a major role in stress response and is known to be epigenetically regulated.
As of 2015 most work has been done in animal models in laboratories, and little work has been done in humans; the work is not yet applicable to clinical psychiatry.[2] Stress-induced epigenetic changes, particularly to genes that effect the hypothalamic–pituitary–adrenal (HPA) axis, persist into future generations, negatively impacting the capacity of offspring to adapt to stress. Early life experiences, even when generations removed, can cause permanent epigenetic modifications of DNA resulting in changes in gene expression, endocrine function and metabolism.[1] These heritable epigenetic modifications include DNA methylation of the promoter regions of genes that affect sensitivity to stress.
Mechanism
Epigenetic modification in response to stress results in molecular and genetic alterations that in turn results in mis-regulated or silenced genes. Heterochromatin is the protein that controls the silencing of these genes epigenetically. For example, epigenetic modifications to the gene BDNF (brain derived neurotrophic factor), as well as dATF-2, as a result of stress can be passed on to offspring. Chronic variable stress induces offspring hypothalamic gene expression modifications, including elevated methylation levels of the BDNF promoter in the hippocampus.[3] This methylation will also occur in the heterochromatin, causing a disrupted heterochromatin to be passed on to the child. Maternal separation and postnatal maternal abuse also increases DNA methylation at regulatory regions of BDNF genes in the prefrontal cortex and hippocampus, leading to potential stress vulnerability in future generations.[4]
Stress can also result in inheritable changes DNA methylation in the promoter regions of the estrogen receptor alpha (ERα),[5] glucocorticoid receptor (GR),[1] and mineralocorticoid receptor (MR).[6] These changes lead to altered expression of these genes in offspring that in turn leads to decreased stress tolerance.
Stress and the HPA axis
Gene regulation as it relates to the HPA axis has been implicated in transgenerational stress effects. Environmental prenatal stress exposure, for example, alters glucocorticoid receptor gene expression, gene function, and future stress response in F1 and F2 generations.[5][1] Maternal care likewise contributes to HPA-related epigenetic modifications. Epigenetic re-programming of gene expression alters stress response in offspring later in life when exposed to decreased maternal care. Inattentive mothering has led to increased levels of gene methyl marks, compared to attentive mothers.[5] Female offspring with low licking-grooming mothers have decreased promoter methylation and increased histone acetylation, leading to increased glucocorticoid receptor expression.[5] Epigenetic modifications as a result of absent maternal care lead to decreased estrogen receptor alpha expression, due to increased methylation at the gene's promoter.[5]
Epigenetic writers, erasers, and readers
Epigenetic changes are performed by enzymes known as writers, which can add epigenetic modifications, erasers, which erase epigenetic modifications, and readers, which can recognize epigenetic modifications and cause a downstream effect. Stress-induced modifications of these writers, erasers, and readers result in important epigenetic modifications such as DNA methylation and acetylation.
DNA methylation

DNA methylation is a type of epigenetic modification in which methyl groups are added to cytosines of DNA. DNA methylation is an important regulator of gene expression and is usually associated with gene repression.
MeCP2
Laboratory studies have found that early life stress in rodents can cause phosphorylation of methyl CpG binding protein 2 (MeCP2), a protein that preferentially binds CpGs and is most often associated with suppression of gene expression. Stress-dependent phosphorylation of MeCP2 causes MeCP2 to dissociate from the promoter region of a gene called arginine vasopressin (avp), causing avp to become demethylated and upregulated. This may be significant because arginine vasopressin is known to regulate mood and cognitive behavior. Additionally, arginine vasopressin upregulates corticotropin-releasing hormone (CRH), which is a hormone important for stress response. Thus, stress-induced upregulation of avp due to demethylation might alter mood, behavior, and stress responses. Demethylation of this locus can be explained by reduced binding of DNA methyl transferases (DNMT), an enzyme that adds methyl groups to DNA, to this locus.[2]
MeCP2 is known to have interactions with several other enzymes that modify chromatin (for example, HDAC-containing complexes and co-repressors) and in turn regulate activity of genes that modulate stress response either by increasing or decreasing stress tolerance. For example, epigenetic upregulation of genes that increase stress response may cause decreased stress tolerance in an organism. These interactions are dependent on the phosphorylation status of MeCP2, which as previously mentioned, can be altered by stress.[2][7]
DNMT1
DNA methyltransferase 1 (DNMT1) belongs to a family of proteins known as DNA methyltransferases, which are enzymes that add methyl groups to DNA. DNMT1 is specifically involved in maintaining DNA methylation; hence it is also known as the maintenance methylase DNMT1. DNMT1 aids in regulation of gene expression by methylating promoter regions of genes, causing transcriptional repression of these genes.
DNMT1 is transcriptionally repressed under stress-mimicking exposure both in vitro and in vivo using a mouse model. Accordingly, transcriptional repression of DNMT1 in response to long-term stress-mimicking exposure causes decreased DNA methylation, which is a marker of gene activation. In particular, there is decreased methylation of a gene called fkbp5, which plays a role in stress response as a glucocorticoid-responsive gene. Thus, chronic stress may cause demethylation and hyperactivation of a stress-related gene, causing increased stress response.[2][8]
Additionally, DNMT1 gene locus has increased methylation in individuals who were exposed to trauma and developed post-traumatic stress disorder (PTSD). Increased methylation of DNMT1 did not occur in trauma-exposed individuals who did not develop PTSD. This may indicate an epigenetic phenotype that can differentiate PTSD-susceptible and PTSD-resilient individuals after exposure to trauma.[2][9]
Transcription factors
Transcription factors are proteins that bind DNA and modulate the transcription of genes into RNA such as mRNA, tRNA, rRNA, and more; thus they are essential components of gene activation. Stress and trauma can affect expression of transcription factors, which in turn alter DNA methylation patterns.
For example, transcription factor nerve growth-induced protein A (NGFI-A, also called NAB1) is up-regulated in response to high maternal care in rodents, and down-regulated in response to low maternal care (a form of early life stress). Decreased NGFI-A due to low maternal care increases methylation of a glucocorticoid receptor promoter in rats. Glucocorticoid is known to play a role in downregulating stress response; therefore, downregulation of glucocorticoid receptor by methylation causes increased sensitivity to stress.[2][10][11]
Histone acetylation
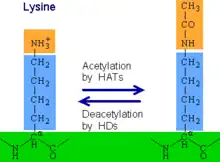
Histone acetylation and deacetylation is a type of epigenetic modification in which acetyl groups are added to lysine on histone tails. Histone acetylation, performed by enzymes known as histone acetyltransferases (HATs), removes the positive charge from lysine and results in gene activation by weakening the histone's interaction with negatively-charged DNA. In contrast, histone deacetylation performed by histone deacetylases (HDACs) results in gene deactivation.
HDAC
Transcriptional activity and expression of HDACs is altered in response to early life stress.[2][12][13] For animals exposed to early life stress, HDAC expression tends to be lower when they are young and higher when they are older. This suggests an age-dependent effect of early life stress on HDAC expression. These HDACs may result in deacetylation and thus activation of genes that upregulate stress response and decrease stress tolerance.[2][14]
Transgenerational epigenetic influences
Genome-wide association studies have shown that psychiatric disorders are partly heritable; however, heritability cannot be fully explained by classical Mendelian genetics. Epigenetics has been postulated to play a role. This is because there is strong evidence of transgenerational epigenetic effects in general. For example, one study found transmission of DNA methylation patterns from fathers to offspring during spermatogenesis. More specifically to mental illnesses, several studies have shown that traits of psychiatric illnesses (such as traits of PTSD and other anxiety disorders) can be transmitted epigenetically. Parental exposure to various stimuli, both positive and negative, can cause these transgenerational epigenetic and behavioral effects.[15]
Parental exposure to trauma and stress
Trauma and stress experienced by a parent can cause epigenetic changes to its offspring. This has been observed both in population and experimental studies.[15]
Human models illustrating transgenerational stress effects are limited due to relatively novel exploration of the topic of epigenetics as well as lengthy follow-up intervals required for multi-generational studies. Several models, however, have investigated the role of epigenetic inheritance and transgenerational stress effects. Transgenerational stress in humans, as in animal models, induces effects influencing social behavior, reproductive success, cognitive ability, and stress response.[3] Similar to animal models, human studies have investigated the role of epigenetics and transgenerational inheritance molecularly as it relates to the HPA system. Prenatal influences, such as emotional stress, nutrition depravation, toxin exposure, hypoxia, increased maternal HPA activity, and cortisol levels may activate or affect HPA axis activity of offspring, despite placental barrier.[16]
Biological vulnerability and HPA axis alterations may be observed after maternal epigenetic programing during pregnancy, leading to similar modifications in future generations.[17] Child abuse exposure, for example, is associated with lower baseline infant cortisol levels as well as modified HPA axis function. Human studies investigating posttraumatic stress disorder (PTSD) and its effects on offspring have illustrated similar molecular and HPA axis modification and function.[6] PTSD patients who experienced trauma from genocides or terrorist attacks frequently exhibited aggressive or neglectful behavior toward offspring during critical developmental periods, possibly contributing to permanent glucocorticoid deregulation in offspring.[17] PTSD mothers and children illustrate lower basal cortisol levels and glucocorticoid receptors and increased mineralocorticoid receptors when exposed to stress.[6] Therefore, developmental experiences, such as stress exposure, may have critical effects on neuromodulatory mechanisms transgenerationally.[16]
Strong relationships between maternal care and subsequent epigenetic modification in offspring, similar to that found in animal models, has been observed in humans. Severe emotional trauma in the mother, for example, often leads to modified methylation patterns of DNA in subsequent offspring generations.[18] PTSD exposed offspring illustrate epigenetic modifications similar to that seen in PTSD mothers, with an increased NR3C2 methylation in exon 1 and increased CpG methylation in the NR3C2 coding sequence, leading to alterations in mineralocorticoid receptor gene expression.[6] Additionally, investigation of post mortem hippocampal tissue indicates decreased levels of neuron-specific glucocorticoid receptor mRNA and decreased DNA methylation in promoter regions among suicidal individuals with lifelong stress and/or abuse exposure.
Epigenetic mechanisms as a result of early life stress may be responsible for neuronal and synaptic alterations in the brain. Developmental stress exposure has been shown to alter brain structure and behavioral functions in adulthood. Evidence of decreased complexity in the CA1 and CA3 region of the hippocampus in terms of dendritic length and spine density after early-life stress exposure indicates transgenerational stress inheritance.[4] Therefore, environmental and experience-dependent synaptic reorganization and structure modifications may lead to increased stress vulnerability and brain dysfunction in future generations.[4]
Paternal stress inheritance
As rodent offspring are fostered mono-parentally and have no direct exposure with their fathers, offspring born of stressed male rodents provide a good model for transgenerational stress inheritance. Direct injection of sperm RNAs to wild type oocytes results in reproducible stress-related modifications.[19] Small non-coding RNAs may serve as a potential mechanism for stress-related genetic changes in offspring. Mouse models of traumatic early life stress exposure result in microRNA modifications and subsequent differences in gene expression and metabolic function.[19] This effect was reproducible by sperm RNA injection, leading to similar gene modifications in future generations. The novelty of this research suggests direct mechanisms capable of altering epigenetics by stress-related factors.
Phenotypic effects
Early life experiences and environmental factors may lead to epigenetic modification at specific gene loci, leading to altered neuronal plasticity, function, and subsequent behavior.[4] Chromatin remodeling in rodent offspring and altered gene expression within the limbic brain regions that may contribute to depression, stress, and anxiety-related disorders in future generations.[4] Variations in maternal care, such as maternal licking and grooming, indicates reduced HPA axis reactivity in subsequent generations.[5] Such HPA axis modifications lead to decreased anxiety-like behavior in adulthood and increased glucocorticoid receptor levels leading to negative feedback on HPA reactivity and further behavioral modifications.[5][1] Rodent models of maternal separation also reveal increased depressive-like behavior in offspring, decreased stress coping abilities, and changes in DNA methylation.[1]
Holocaust
An epidemiological study investigating behavioral, physiological, and molecular changes in the children of Holocaust survivors found epigenetic modifications of a glucocorticoid receptor gene, Nr3c1. This is significant because glucocorticoid is a regulator of the hypothalamus-pituitary-adrenal axis (HPA) and is known to affect stress response. These stress-related epigenetic changes were accompanied by other characteristics that indicated higher stress and anxiety in these offspring, including increased symptoms of PTSD, greater risk of anxiety, and higher levels of the stress hormone cortisol.[15][20][21][22]
Experimental evidence
The effect of parental exposure to stress has been tested experimentally as well. For example, male mice who were put under early life stress through poor maternal care—a scenario analogous to human childhood trauma—passed on epigenetic changes that resulted in behavioral changes in offspring. Offspring experienced altered DNA methylation of stress-response genes such as CB1 and CRF2 in the cortex, as well as epigenetic alterations in transcriptional regulation gene MeCP2. Offspring were also more sensitive to stress, which is in accordance with the altered epigenetic profile. These changes persisted for up to three generations.[15][23]
In another example, male mice were socially isolated as a form of stress. Offspring of these mice had increased anxiety in response to stressful conditions, increased stress hormone levels, dysregulation of the HPA axis which plays a key role in stress response, and several other characteristics that indicated increased sensitivity to stress.[15][23]
Inheritance of small-noncoding RNA
Studies have found that early life stress induced through poor maternal care alters sperm epigenome in male mice. In particular, expression patterns of small-noncoding RNAs (sncRNAs) are altered in the sperm, as well as in stress-related regions of the brain.[24][25][26] Offspring of these mice exhibited the same sncRNA expression changes in the brain, but not in the sperm. These changes were coupled with behavioral changes in the offspring that were comparable to behavior of the stressed fathers, especially in terms of stress response. Additionally, when the sncRNAs in the fathers' sperm were isolated and injected into fertilized eggs, the resulting offspring inherited the stress behavior of the father. This suggests that stress-induced modifications of sncRNAs in sperm can cause inheritance of stress phenotype independent of the father's DNA.[24][25]
Exercise
Just as parental stress can alter epigenetics of offspring, parental exposure to positive environmental factors cause epigenetic modifications as well. For example, male mice that participated in voluntary physical exercise resulted in offspring that had reduced fear memory and anxiety-like behavior in response to stress. This behavioral change likely occurred due to expressions of small non-coding RNAs that were altered in sperm cells of the fathers.[15][27]
Stress effect reversal
Additionally, exposing fathers to enriching environments can reverse the effect of early life stress on their offspring. When early life stress is followed by environmental enrichment, anxiety-like behavior in offspring is prevented.[15][28] Similar studies have been conducted in humans and suggest that DNA methylation plays a role.[29]
Post-traumatic stress disorder (PTSD)
Post-traumatic stress disorder (PTSD) is a stress-related mental health disorder that emerges in response to traumatic or highly stressful experiences. It is believed that PTSD develops as a result of an interaction between these traumatic experiences and genetic factors. The signs and symptoms of PTSD can include avoidance behaviors, invasive thoughts, and significant alterations in normal behavior and thinking.[30] There is evidence suggesting PTSD formation is associated with epigenetic changes such as DNA methylation and acetylation of histone proteins. Increased DNA methylation has been found to regulate the induction of fear conditioning behaviors associated with PTSD triggers.[31][9] Histone modifications, like acetylation and deacetylation, play an important role in the development of PTSD, which is related to fear memory from traumatic events.[32]
The DSM-5 asserts that PTSD manifests differently in children over six years old than in adults. Specifically that their flashbacks and/or intrusive memories may be explained by recreating their traumatic event(s) through their play. They may also experience reoccurring nightmares that are indirectly related to the event. Additionally, there is a separate criteria altogether for PTSD in children under six years old.[33]
Epigenetic Modifications
DNA methylation
Through a number of human studies, PTSD is known to affect DNA methylation of CpG islands in several genes involved in numerous activities, including stress responses and neurotransmitter activity. CpGs are used to describe cytosine-guanine adjacent nucleotides within the same strand of DNA. CpG islands are defined by computer algorithms as being made up of at least 60% CpGs and being anywhere between 200 and 3000 base pairs in size. The methylation of these CpG islands can cause histone modifications which can lead to the condensation of chromatin which can ultimately alter gene expression.[30]
DNMT Enzyme
DNA methyltransferase, DNMT, is an enzyme responsible for increased methylation of DNA. It has been found that DNMT and its associated increased methylation can regulate risk for memory consolidation and fear conditioning.[31]
TET Enzyme
The removal of methyl groups from cytosine is initiated by a TET enzyme. TET is an enzyme known to oxidize 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC) within the genome. This reaction initiates active DNA demethylation to ultimately alter gene expression. It has been found that the TET enzyme exists as two isoforms which are differentially regulated and expressed across brain regions. The regulation of these isoforms can affect synaptic connections and ultimately memory formation.[34] The manipulation of the TET enzymes' expression levels has become a potential source of interest for PTSD medication.[31][9]
The table below identifies differentially methylated regions (DMRs) across the genome which undergo PTSD-induced epigenetic changes which alter gene expression.
| Genetic Loci | Finding(s) |
|---|---|
| SLC6A4 | Following trauma exposure, low methylation levels of SLC6A4 increases risk of PTSD; high methylation levels decreases risk of PTSD[36] |
| MAN2C1 | Higher MAN2C1 methylation is correlated to greater risk of PTSD in individuals exposed to traumatic events[37] |
| TPR, CLEC9A, APC5, ANXA2, TLR8 | PTSD is associated with increased global methylation of these genes[38] |
| ADCYAP1R1 | Higher methylation is associated with PTSD symptoms in individuals exposed to trauma[39] |
| LINE-1, Alu | Higher methylation of these loci is observed in postdeployed veterans who developed PTSD compared to those who do not develop PTSD[40] |
| SLC6A3 | High SLC6A3 promoter methylation, combined with a nine-repeat allele of SLC6A3, is correlated to higher PTSD risk[41] |
| IGF2, H19, IL8, IL16, IL18 | Higher methylation of IL18 but lower methylation of H19 and IL18 is associated with deployed veterans who developed PTSD<[42] |
| NR3C1 | Lower methylation levels of NR3C1 1B and 1C promoters is associated with PTSD;[43]
Fathers with PTSD have offspring with higher NR3C1 1F promoter methylation;[22] Lower levels of NR3C1 1F promoter methylation is associated with PTSD in combat veterans;[44] Higher levels of NR3C1 methylation in male (but not female) Rwandan genocide survivors is associated with decreased PTSD risk[45] |
| SPATC1L | Higher methylation is associated with PTSD symptoms. |
| HLA-DPB1 | Higher methylation is associated with PTSD symptoms. |
Histone Modifications
Histone acetylation is performed by histone acetyl transferases (HATs) and histone deacetylation is carried out by histone deacetylases (HDACs).[46] In rodent PTSD models, it has been found that an increase in histone acetylation is associated with fear conditioning.[46] Histone acetylation can be involved in all parts of fear memory, including the development to memory extinction. It can also play a role in long-term potentiation (LTP).[35] It was also observed that HDACs increase memory formation in fear extinction and HDAC inhibitors (HDACi) have shown evidence for modifying memory extinction, a possible treatment for PTSD.[46]
Nervous System Structures Affected by PTSD
Hypothalamus-pituitary-adrenal axis
The hypothalamus-pituitary-adrenal (HPA) axis is a neuroendocrine system largely involved in ascertaining the levels of cortisol circulating the body at any given point in time. As cortisol plays a key role in the stress response, so does the HPA axis. The dysregulation of the HPA axis has been found to be characteristic of several stress disorders, including PTSD. This system works under a negative feedback loop structure. Hence, this HPA axis dysregulation may take the form of amplified negative inhibition and result in down-regulated cortisol levels.[47] Epigenetic modifications play a role in this dysregulation, and these modifications are likely caused by the traumatic/stressful experience that triggered PTSD.[35]
Immune dysregulation by HPA Axis Modifications
PTSD is often linked with immune dysregulation. Traumatic experiences can induce epigenetic changes at the gene loci that are immune-related which can lead to immune dysregulation and an increased risk of PTSD.[35] Trauma exposure can also disrupt the HPA axis, thus altering peripheral immune function. The effect of PTSD on immune function arises in at least two ways: 1) Continuous disturbances on the HPA axis can dysregulate peripheral immune function, and 2) the effects of immune dysregulation in the periphery can lead to increased development of PTSD because of alterations in brain function.[35]
PTSD-associated changes in immune cells found in blood or saliva can serve as biomarkers that trigger epigenetic changes which are involved in the pathogenesis of PTSD.[35] These unique biomarkers serve as means of identifying PTSD subtypes. Beyond identifying subtypes, these distinct biomarkers can potentially be used to develop PTSD treatments.[35]
Epigenetic modifications have been observed in immune-related genes of individuals with PTSD. For example, deployed military members who developed PTSD have higher methylation in the immune-related gene interleukin-18 (IL-18).[35][42] This has interested scientists because high levels of IL-18 increase cardiovascular disease risk, and individuals with PTSD have elevated cardiovascular disease risk. Thus, stress-induced immune dysregulation via methylation of IL-18 may play a role in cardiovascular disease in individuals with PTSD.[35][48][38]
Additionally, an epigenome-wide study found that individuals with PTSD have altered levels of methylation in the following immune-related genes: TPR, CLEC9A, APC5, ANXA2, TLR8, IL-4, and IL-2. This again shows that immune function in PTSD is disrupted, especially by epigenetic changes that are likely stress-induced.[35][38]
Genes Affected by PTSD
NR3C1
Nr3c1 is a transcription factor that encodes a glucocorticoid receptor (GR) and contains many GR response elements. Npas4 is another regulatory transcription factor also responsible for the regulation of GRs. Stress-induced changes in Nr3c1 and Npas4 methylation have been shown to alter stress sensitivity. This response differs between short-lived stress exposure and chronic stress exposure. In response to short lived stress, the NR3C1 promoter is more hydroxymethylated which is a modification associated with increased transcription of GR-associated genes. Thus, short lived stress exposure increases stress sensitivity. Conversely, in response to chronic stress, the Npas4 promoter has been presumed to be increased in methylation, a modification which is associated with inhibitory regulation of GRs. Thus, chronic stress exposure decreases stress sensitivity. These distinctions are important in understanding the epigenetic patterns of stress and genetic interactions with PTSD triggers.[49] Overall, in the hippocampus of chronically stressed animals, the 3′-UTR (untranslated region of DNA) of the glucocorticoid receptor Nr3c1 showed increased hydroxymethylation, which led to increased transcription and thus, the disruption of stress tolerance and increased risk of disorders such as PTSD. However, early life stress increases methylation of the 1F promoter in this gene (or the 17 promoter analog in rodents). Because of its role in stress response and its link to early life stress, this gene has been of particular interest in the context of PTSD and has been studied in PTSD of both combat veterans and civilians.[35]
In studies involving combat veterans, those who developed PTSD had lowered methylation of the Nr3c1 1F promoter compared to those who did not develop PTSD. Additionally, veterans who developed PTSD and had higher Nr3c1 promoter methylation responded better to long-term psychotherapy compared to veterans with PTSD who had lower methylation. These findings were recapitulated in studies involving civilians with PTSD.[35][50] In civilians, PTSD is linked to lower methylation levels in the T-cells of exons 1B and 1C of Nr3c1, as well as higher GR expression. Thus, it seems that PTSD causes lowered methylation levels of GR loci and increased GR expression.[35][43] The methylation of GR in T-cells are investigated because of its role in regulating cell immunity which, as such, stores cellular memory with environmental factors. T-cell fragments from individual cell populations are preferred over homogenized tissue because of the drastic variation in DNA methylation patterns between different cell fragments.
Although these results of decreased methylation and hyperactivation of GR conflict with the effect of early life stress at the same loci, these results match previous findings that distinguish HPA activity in early life stress versus PTSD. For example, cortisol levels of HPA in response to early life stress is hyperactive, whereas it is hypoactive in PTSD. Thus, the timing of trauma and stress—whether early or later in life—can cause differing effects on HPA and GR.[43]
FKBP5
Fkbp5 encodes a GR-responsive protein known as Fk506 binding protein 51 (FKBP5). FKBP5 is induced by GR activation and functions in negative feedback by binding GR and reducing GR signaling.[35][51][52][53] There is particular interest in this gene because some FKBP5 alleles have been correlated with increased risk of PTSD and development of PTSD symptoms—especially in PTSD caused by early life adversity. Therefore, FKBP5 likely plays an important role in PTSD.[35][54][55][56]
As mentioned previously, certain FKBP5 alleles are correlated to increase PTSD risk, especially due to early life trauma. It is now known that epigenetic regulation of these alleles is also an important factor. For example, CpG sites in intron 7 of FKBP5 are demethylated after exposure to childhood trauma, but not adult trauma.[35][57] Additionally, methylation of FKBP5 is alters in response to PTSD treatment; thus methylation levels of FKBP5 might correspond to PTSD disease progression and recovery.[35][22]
ADCYAP1 and ADCYAP1R1
Pituitary adenylate cyclase-activating polypeptide (ADCYAP1) and its receptor (ADCYAP1R1) are stress responsive genes that play a role in modulating stress, among many other functions. Additionally, high levels of ADCYAP1 in peripheral blood is correlated to PTSD diagnosis in females who have experienced trauma, thus making ADCYAP1 a gene of interest in the context of PTSD.[35]
Epigenetic regulation of these loci in relation to PTSD still require further investigation, but one study has found that high methylation levels of CpG islands in ADCYAP1R1 can predict PTSD symptoms in both males and females.[35][39]
Alcohol use disorder
Alcohol dependence and stress interact in many ways. For example, stress-related disorders such as anxiety and PTSD are known to increase risk of alcohol use disorder (AUD), and they are often co-morbid. This may in part be due to the fact that alcohol can alleviate some symptoms of these disorders, thus promoting dependence on alcohol. Conversely, early exposure to alcohol can increase vulnerability to stress and stress-related disorders. Moreover, alcohol dependence and stress are known to follow similar neuronal pathways, and these pathways are often dysregulated by similar epigenetic modifications.[58]
HDAC
Histone acetylation is dysregulated by alcohol exposure and dependence, often through dysregulated expression and activity of HDACs, which modulate histone acetylation by removing acetyl groups from lysines of histone tails. For example, HDAC expression is upregulated in chronic alcohol use models.[58] Monocyte-derived dendritic cells of alcohol users have increased HDAC gene expression compared to non-users.[59] These results are also supported by in vivo rat studies, which show that HDAC expression is higher in alcohol-dependent mice that in non-dependent mice. Furthermore, knockout of HDAC2 in mice helps lower alcohol dependence behaviors.[60] The same pattern of HDAC expression is seen in alcohol withdrawal, but acute alcohol exposure has the opposite effect; in vivo, HDAC expression and histone acetylation markers are decreased in the amygdala.[60]
Dysregulation of HDACs is significant because it can cause upregulation or downregulation of genes that have important downstream effects both in alcohol dependence and anxiety-like behaviors, and the interaction between the two. A key example is BDNF (see "BDNF" below).
BDNF
Brain-derived neurotrophic factor (BDNF) is a key protein that is dysregulated by HDAC dysregulation. BDNF is a protein that regulates the structure and function of neuronal synapses. It plays an important role in neuronal activation, synaptic plasticity, and dendritic morphology—all of which are factors that may affect cognitive function. Dysregulation of BDNF is seen both in stress-related disorders and alcoholism; thus BDNF is likely an important molecule in the interaction between stress and alcoholism.[58][61]
For example, BDNF is dysregulated by acute ethanol exposure. Acute ethanol exposure causes phosphorylation of CREB, which can cause increased histone acetylation at BDNF loci. Histone acetylation upregulates BDNF, in turn upregulating a downstream BDNF target called activity-regulated cytoskeleton associated protein (Arc), which is a protein responsible for dendritic spine structure and formation. This is significant because activation of Arc can be associated with anxiolytic (anxiety-reducing) effects. Therefore, ethanol consumption can cause epigenetic changes that alleviate stress and anxiety, thereby creating a pattern of stress-induced alcohol dependence.[58][61][62]
Alcohol dependence is exacerbated by ethanol withdrawal. This is because ethanol withdrawal has the opposite effect of ethanol exposure; it causes lowered CREB phosphorylation, lowered acetylation, downregulation of BDNF, and increase in anxiety. Consequently, ethanol withdrawal reinforces desire for anxiolytic effects of ethanol exposure. Moreover, it is proposed that chronic ethanol exposure results in upregulation of HDAC activity, causing anxiety-like effects that can no longer be alleviated by acute ethanol exposure.[58][61][60]
Potential epigenetic drug treatments
The most common treatments for anxiety disorders at the moment are benzodiazepines, Buspirone, and antidepresants. However, around one/third of patients with anxiety disorders do not respond well to the current anxiolytics, and many others have treatment-resistant anxiety disorders.[63][64] Recent research surrounding DNA methylation changes in genes in genes encoding proteins associated with the HPA axis, histone modifications, and sncRNAs point to epigenetic drugs potentially being effective treatment methods for anxiety disorders.[65]
HDACi
Histone deacetylase inhibitors (HDACi) fall into five different classes, not to be confused with the four different classes of HDACs. The five classes of HDACi consist of (I) hydroxamic acids, (II) short-chain fatty acids, (III) benzamides, (IV) cyclic tetrapeptides, and (V) sirtuin inhibitors. The three classes of HDACs are class I, consisting of HDAC1, HDAC2, HDAC3, HDAC8, class II, consisting of HDAC4, HDAC5, HDAC6, HDAC7, HDAC9, HDAC10, class III, consisting of NAD+-dependent HDACS, and class IV, consisting of HDAC11. While most HDACi inhibit only specific classes of HDACs, certain HDACi can act against all classes, making them pan-inhibitors.[66][67]
HDACi are currently being researched as potential anxiolytics. At the moment, the mechanism of action of HDAC inhibitors in the treatment of anxiety disorders is not clear, as they affect several targets and have multiple pharmacological effects besides the inhibition of HDACs. However, they have been shown to cause DNA demethylation, possibly due to an increase in the levels of TET1, which is a demethylating enzyme.[68] In the human peripheral cells of patients with anxiety disorders and in animal models of anxiety disorders, genes such as GAD1, NR3C1, BDNF, MAOA, HECA, and FKBP5 are shown to be hypermethylated. As such, the mechanism of action of HDACi in anxiety disorders could, in part, be potentially explained by the demethylation of those genes.
Valproate
Valproate is a drug that acts as an HDACi on class I and II HDACs. Six clinical trials surrounding its use as an anxiolytic have been performed so far. Five of the six trials were performed on patients with anxiety disorders, and one of the trials used healthy subjects with no anxiety disorders. Of the five trials performed on patients with anxiety disorders, three found that Valproate decreases panic disorder, one found that Valproate decreases social anxiety, and one found that Valproate reduces generalized anxiety.[69][70][71][72][73] The trial performed on healthy subjects found that Valproate reduces anxiety and also acts as a nerve conduction inhibitor, which could be an explanation for some of its anxiety-reducing effects.[74]
D-cycloserine, Trichostatin-a, Suberoylanilide hydroxamic acid, sodium butyrate, and valproic acid
Various preclinical drug trials using other HDAC inhibitors have also been performed, with most drugs targeting HDAC classes I and II and a select few targeting classes IV and III. The HDACi drug, d-cycloserine, was found to reduce fear in 129S1/SvImJ mice, which are mice that show poor extinction acquisition and recovery of fear-induced suppression of heart-rate variability, enlarged dendritic arbors in basolateral amygdala neurons, and functional abnormalities in cortico-amygdala circuitry that mediates fear extinction.[75] Trichostatin-a was normalized BDNF and Arc expression in the central and the medial nucleus of the amygdala in rats experiencing alcohol withdrawal.[76] Suberoylanilide hydroxamic acid significantly reversed anxiety-like behaviors and stress-induced gastrointestinal hypersensitivity and fecal pellet output.[77] Anxiety-like and depression-like behaviors caused by immobilization stress and/or nicotine addiction were also reduced in mice treated with the HDACi sodium butyrate and valproic acid.[78]
Lactate
Lactate, a metabolite that is naturally produced during exercise, was found to function as an HDAC II and III modulator in a pre-clinical trial. The trial was performed on C57BL/6 mice, which are mice that were exposed to chronic stress in the form of daily defeat by a CD-1 aggressive mouse. While control mice exhibited increased social avoidance, anxiety, and susceptibility to depression, mice that received lactate before each defeat demonstrated resilience to depression and stress and reduced social avoidance and anxiety. Lactate promoted this resilience by restoring regular hippocampal class I HDAC levels and activity.[79]
sncRNA
Preliminary research has been done about therapy involving small non-coding RNAs, demonstrating that they can regulate epigenetic mechanisms of gene expression and could present as biomarkers for disease. One therapy option is for the sncRNAs in patients with anxiety disorders to be targeted for upregulation. Another option is to inhibit the miRNAs in order to reduce their effects, potentially using antisense oligonucleotides or antagomirs as inhibitors.[65]
Hydrocortisone
The medication hydrocortisone is a synthetic form of cortisol. In recent years, the administration of hydrocortisone has been tested as a possible preventative measure for the onset of PTSD symptoms. Ideally, it should be administered immediately after a traumatic event. The efficacy of hydrocortisone as a preventative intervention for PTSD has been confirmed by a meta-analysis of eight separate studies, and researchers believe the best results are obtained when hydrocortisone is administered within the first six hours of exposure to the traumatic event. At this time, however, no curative properties have been discovered.[80] Hydrocortisone's potential operates on two bases: restoration of normal HPA axis functioning and interference with memory consolidation.
HPA Axis Homeostasis
Our standard understandings of PTSD may suggest elevated glucocorticoid levels during and directly following events of trauma. However, multiple studies have indicated that overall HPA axis activity and cortisol levels are depleted in the critical aftermath and extended period after trauma.[81] Moreover, research has also indicated that an appropriate release of glucocorticoids following acute stress may restore homeostatic equilibrium of the HPA axis, thereby preventing gradual sensitization, which is responsible for persistent cortisol reduction and increased PTSD susceptibility. Thus, the appropriately-dosed administration of hydrocortisone promptly following the traumatic incident would normalize the HPA axis and potentially prevent PTSD onset.[80]
Disruption of Memory Consolidation
In the absence of memory reactivation, hydrocortisone's effectiveness within a six-hour window supports the consolidation theory, which asserts that memory is labile even immediately after trauma. It is assumed that the medication is disrupting initial memory consolidation of the traumatic event. However, its exact mechanism within this context remains largely unknown.
Although trials have proven promising, there is much more research to be done. Further comprehensive studies are required amidst more diverse populations under different traumatic conditions in order to ascertain factors of optimal usage of the drug and clarify the PTSD subgroups hydrocortisone is beneficial to.
Rodent Models Used to Study PTSD Medication
Stress-enhanced fear learning (SEFL)
The observation of epigenetic modifications and their role in regulating fear learning is an active area of research. The use of stress-enhanced fear learning (SEFL) paradigms are important for forming preclinical models of PTSD because one is able to observe the epigenetic changes in rodents and PTSD associated changes in fear learning after stress exposure.[49]
Single-prolonged stress (SPS)
The single-prolonged stress (SPS) model is a tool in which a complex stressor is consistently presented. This tool is used to explore the complexity of PTSD, particularly its impaired fear extinction.[49]
Susceptible Factors to PTSD
Despite high levels of individuals exposed to trauma, only about one third of exposed individuals develop PTSD. This suggest that individuals differ in their susceptibility to PTSD. This might arise from differences in the epigenetic modifications that they generate in response to traumatic experiences. Furthermore, a large area of research regarding increased susceptibility to PTSD investigates the transgenerational inheritance of epigenetic modifications resulting from trauma.[82] A recent review of PTSD susceptibility suggested that a range as wide as 30% to 70% of susceptibility to PTSD can be attributed to heritability.[83] From our observations of mice in transgenerational research, we have seen that the epigenetic modifications that stem from trauma can be passed down multiple generations.[82] Epigenetic modifications due trauma are not the only heritable factors that affect PTSD susceptibility. General histories of health deficiencies, both physical and psychological, have also been associated with a higher PTSD susceptibility. Sociodemographic factors may come into play as well. Particularly, ethnic minorities and women are more susceptible to development of PTSD.[84]
See also
References
- Matthews SG, Phillips DI (January 2012). "Transgenerational inheritance of stress pathology". review. Experimental Neurology. 233 (1): 95–101. doi:10.1016/j.expneurol.2011.01.009. PMID 21281632. S2CID 30261742.
- Klengel T, Binder EB (June 2015). "Epigenetics of Stress-Related Psychiatric Disorders and Gene × Environment Interactions". review. Neuron. 86 (6): 1343–57. doi:10.1016/j.neuron.2015.05.036. PMID 26087162.

- Gudsnuk K, Champagne FA (2012). "Epigenetic influence of stress and the social environment". review. ILAR Journal. 53 (3–4): 279–88. doi:10.1093/ilar.53.3-4.279. PMC 4021821. PMID 23744967.
- Vialou V, Feng J, Robison AJ, Nestler EJ (2013). "Epigenetic mechanisms of depression and antidepressant action". review. Annual Review of Pharmacology and Toxicology. 53: 59–87. doi:10.1146/annurev-pharmtox-010611-134540. PMC 3711377. PMID 23020296.
- Szyf M, Weaver I, Meaney M (July 2007). "Maternal care, the epigenome and phenotypic differences in behavior". review. Reproductive Toxicology. 24 (1): 9–19. doi:10.1016/j.reprotox.2007.05.001. PMID 17561370.
- Daskalakis NP, Cohen H, Nievergelt CM, Baker DG, Buxbaum JD, Russo SJ, Yehuda R (October 2016). "New translational perspectives for blood-based biomarkers of PTSD: From glucocorticoid to immune mediators of stress susceptibility". review. Experimental Neurology. 284 (Pt B): 133–140. doi:10.1016/j.expneurol.2016.07.024. PMC 5056828. PMID 27481726.
- Bellini E, Pavesi G, Barbiero I, Bergo A, Chandola C, Nawaz MS, Rusconi L, Stefanelli G, Strollo M, Valente MM, Kilstrup-Nielsen C, Landsberger N (2014). "MeCP2 post-translational modifications: a mechanism to control its involvement in synaptic plasticity and homeostasis?". review. Frontiers in Cellular Neuroscience. 8: 236. doi:10.3389/fncel.2014.00236. PMC 4131190. PMID 25165434.
- Lee RS, Tamashiro KL, Yang X, Purcell RH, Huo Y, Rongione M, Potash JB, Wand GS (November 2011). "A measure of glucocorticoid load provided by DNA methylation of Fkbp5 in mice". primary. Psychopharmacology. 218 (1): 303–12. doi:10.1007/s00213-011-2307-3. PMC 3918452. PMID 21509501.
- Sipahi L, Wildman DE, Aiello AE, Koenen KC, Galea S, Abbas A, Uddin M (November 2014). "Longitudinal epigenetic variation of DNA methyltransferase genes is associated with vulnerability to post-traumatic stress disorder". primary. Psychological Medicine. 44 (15): 3165–79. doi:10.1017/S0033291714000968. PMC 4530981. PMID 25065861.
- Weaver IC, Cervoni N, Champagne FA, D'Alessio AC, Sharma S, Seckl JR, Dymov S, Szyf M, Meaney MJ (August 2004). "Epigenetic programming by maternal behavior". primary. Nature Neuroscience. 7 (8): 847–54. doi:10.1038/nn1276. PMID 15220929. S2CID 1649281.
- Zhang TY, Labonté B, Wen XL, Turecki G, Meaney MJ (January 2013). "Epigenetic mechanisms for the early environmental regulation of hippocampal glucocorticoid receptor gene expression in rodents and humans". review. Neuropsychopharmacology. 38 (1): 111–23. doi:10.1038/npp.2012.149. PMC 3521971. PMID 22968814.
- Levine A, Worrell TR, Zimnisky R, Schmauss C (January 2012). "Early life stress triggers sustained changes in histone deacetylase expression and histone H4 modifications that alter responsiveness to adolescent antidepressant treatment". primary. Neurobiology of Disease. 45 (1): 488–98. doi:10.1016/j.nbd.2011.09.005. PMC 3225638. PMID 21964251.
- Tesone-Coelho C, Morel LJ, Bhatt J, Estevez L, Naudon L, Giros B, Zwiller J, Daugé V (January 2015). "Vulnerability to opiate intake in maternally deprived rats: implication of MeCP2 and of histone acetylation". primary. Addiction Biology. 20 (1): 120–31. doi:10.1111/adb.12084. PMID 23980619. S2CID 5938827.
- Suri D, Bhattacharya A, Vaidya VA (February 2014). "Early stress evokes temporally distinct consequences on the hippocampal transcriptome, anxiety and cognitive behaviour". primary. The International Journal of Neuropsychopharmacology. 17 (2): 289–301. doi:10.1017/S1461145713001004. PMID 24025219.
- Yeshurun S, Hannan AJ (March 2018). "Transgenerational epigenetic influences of paternal environmental exposures on brain function and predisposition to psychiatric disorders". review. Molecular Psychiatry. 24 (4): 536–548. doi:10.1038/s41380-018-0039-z. PMID 29520039. S2CID 3767345.
- Li SC (November 2013). "Neuromodulation and developmental contextual influences on neural and cognitive plasticity across the lifespan". review. Neuroscience and Biobehavioral Reviews. 37 (9 Pt B): 2201–8. doi:10.1016/j.neubiorev.2013.07.019. PMID 23973556. S2CID 46501356.
- Yahyavi ST, Zarghami M, Marwah U (2013). "A review on the evidence of transgenerational transmission of posttraumatic stress disorder vulnerability". review. Revista Brasileira de Psiquiatria. 36 (1): 89–94. doi:10.1590/1516-4446-2012-0995. PMID 24402183.
- Kellermann NP (2013). "Epigenetic transmission of Holocaust trauma: can nightmares be inherited?". review. The Israel Journal of Psychiatry and Related Sciences. 50 (1): 33–9. PMID 24029109.
- Hodes GE, Walker DM, Labonté B, Nestler EJ, Russo SJ (January 2017). "Understanding the epigenetic basis of sex differences in depression". review. Journal of Neuroscience Research. 95 (1–2): 692–702. doi:10.1002/jnr.23876. PMC 5130105. PMID 27870456.
- Yehuda R, Halligan SL, Bierer LM (2001). "Relationship of parental trauma exposure and PTSD to PTSD, depressive and anxiety disorders in offspring". primary. Journal of Psychiatric Research. 35 (5): 261–70. doi:10.1016/S0022-3956(01)00032-2. PMID 11591428.
- Yehuda R, Bierer LM (2008). "Transgenerational transmission of cortisol and PTSD risk". Stress Hormones and Post Traumatic Stress Disorder Basic Studies and Clinical Perspectives. review. Progress in Brain Research. Vol. 167. pp. 121–35. doi:10.1016/S0079-6123(07)67009-5. ISBN 9780444531407. PMID 18037011.
- Yehuda R, Daskalakis NP, Lehrner A, Desarnaud F, Bader HN, Makotkine I, Flory JD, Bierer LM, Meaney MJ (August 2014). "Influences of maternal and paternal PTSD on epigenetic regulation of the glucocorticoid receptor gene in Holocaust survivor offspring". primary. The American Journal of Psychiatry. 171 (8): 872–880. doi:10.1176/appi.ajp.2014.13121571. PMC 4127390. PMID 24832930.
- Franklin TB, Russig H, Weiss IC, Gräff J, Linder N, Michalon A, Vizi S, Mansuy IM (September 2010). "Epigenetic transmission of the impact of early stress across generations". primary. Biological Psychiatry. 68 (5): 408–15. doi:10.1016/j.biopsych.2010.05.036. PMID 20673872. S2CID 15851484.
- Yuan TF, Li A, Sun X, Ouyang H, Campos C, Rocha NB, Arias-Carrión O, Machado S, Hou G, So KF (November 2016). "Transgenerational Inheritance of Paternal Neurobehavioral Phenotypes: Stress, Addiction, Ageing and Metabolism". review. Molecular Neurobiology. 53 (9): 6367–6376. doi:10.1007/s12035-015-9526-2. hdl:10400.22/7331. PMID 26572641. S2CID 25694221.
- Gapp K, Jawaid A, Sarkies P, Bohacek J, Pelczar P, Prados J, Farinelli L, Miska E, Mansuy IM (May 2014). "Implication of sperm RNAs in transgenerational inheritance of the effects of early trauma in mice". primary. Nature Neuroscience. 17 (5): 667–9. doi:10.1038/nn.3695. PMC 4333222. PMID 24728267.
- Sharma U, Rando OJ (June 2014). "Father-son chats: inheriting stress through sperm RNA". comment. Cell Metabolism. 19 (6): 894–5. doi:10.1016/j.cmet.2014.05.015. PMC 4131544. PMID 24896534.
- Short AK, Yeshurun S, Powell R, Perreau VM, Fox A, Kim JH, Pang TY, Hannan AJ (May 2017). "Exercise alters mouse sperm small noncoding RNAs and induces a transgenerational modification of male offspring conditioned fear and anxiety". primary. Translational Psychiatry. 7 (5): e1114. doi:10.1038/tp.2017.82. PMC 5534950. PMID 28463242.
- Gapp K, Bohacek J, Grossmann J, Brunner AM, Manuella F, Nanni P, Mansuy IM (October 2016). "Potential of Environmental Enrichment to Prevent Transgenerational Effects of Paternal Trauma". primary. Neuropsychopharmacology. 41 (11): 2749–58. doi:10.1038/npp.2016.87. PMC 5026744. PMID 27277118.
- Mitchell C, Schneper LM, Notterman DA (January 2016). "DNA methylation, early life environment, and health outcomes". review. Pediatric Research. 79 (1–2): 212–9. doi:10.1038/pr.2015.193. PMC 4798238. PMID 26466079.
- Katrinli, Seyma; Zheng, Yuanchao; Gautam, Aarti; Hammamieh, Rasha; Yang, Ruoting; Venkateswaran, Suresh; Kilaru, Varun; Lori, Adriana; Hinrichs, Rebecca; Powers, Abigail; Gillespie, Charles F. (January 2021). "PTSD is associated with increased DNA methylation across regions of HLA-DPB1 and SPATC1L". Brain, Behavior, and Immunity. 91: 429–436. doi:10.1016/j.bbi.2020.10.023. ISSN 1090-2139. PMC 7749859. PMID 33152445.
- "Whitehead Institute of MIT". Whitehead Institute of MIT. Retrieved 2022-05-20.
- Zhang, Yingqian; Zhao, Guangyi; Han, Yuwei; Zhang, Jingyi; Cao, Chengqi; Wang, Li; Zhang, Kunlin (2022). "The mechanisms of histone modification in post-traumatic stress disorder". Advances in Psychological Science. 30 (1): 98. doi:10.3724/SP.J.1042.2022.00098. ISSN 1671-3710.
- American Psychiatric Association (2013). Diagnostic and statistical manual of mental disorders (5th ed.). Arlington. p. 271.
- Greer, C. B.; Wright, J.; Weiss, J. D.; Lazarenko, R. M.; Moran, S. P.; Zhu, J.; Chronister, K. S.; Jin, A. Y.; Kennedy, A. J.; Sweatt, J. D.; Kaas, G. A. (2021-01-27). "Tet1 Isoforms Differentially Regulate Gene Expression, Synaptic Transmission, and Memory in the Mammalian Brain". The Journal of Neuroscience. 41 (4): 578–593. doi:10.1523/JNEUROSCI.1821-20.2020. ISSN 1529-2401. PMC 7842754. PMID 33262245.
- Zannas AS, Provençal N, Binder EB (September 2015). "Epigenetics of Posttraumatic Stress Disorder: Current Evidence, Challenges, and Future Directions". review. Biological Psychiatry. 78 (5): 327–35. doi:10.1016/j.biopsych.2015.04.003. PMID 25979620. S2CID 20876167.
- Koenen KC, Uddin M, Chang SC, Aiello AE, Wildman DE, Goldmann E, Galea S (August 2011). "SLC6A4 methylation modifies the effect of the number of traumatic events on risk for posttraumatic stress disorder". review. Depression and Anxiety. 28 (8): 639–47. doi:10.1002/da.20825. PMC 3145829. PMID 21608084.
- Uddin M, Galea S, Chang SC, Aiello AE, Wildman DE, de los Santos R, Koenen KC (2011). "Gene expression and methylation signatures of MAN2C1 are associated with PTSD". primary. Disease Markers. 30 (2–3): 111–21. doi:10.1155/2011/513659. PMC 3188659. PMID 21508515.
- Smith AK, Conneely KN, Kilaru V, Mercer KB, Weiss TE, Bradley B, Tang Y, Gillespie CF, Cubells JF, Ressler KJ (September 2011). "Differential immune system DNA methylation and cytokine regulation in post-traumatic stress disorder". primary. American Journal of Medical Genetics. Part B, Neuropsychiatric Genetics. 156B (6): 700–8. doi:10.1002/ajmg.b.31212. PMC 3292872. PMID 21714072.
- Ressler KJ, Mercer KB, Bradley B, Jovanovic T, Mahan A, Kerley K, Norrholm SD, Kilaru V, Smith AK, Myers AJ, Ramirez M, Engel A, Hammack SE, Toufexis D, Braas KM, Binder EB, May V (February 2011). "Post-traumatic stress disorder is associated with PACAP and the PAC1 receptor". primary. Nature. 470 (7335): 492–7. Bibcode:2011Natur.470..492R. doi:10.1038/nature09856. PMC 3046811. PMID 21350482.
- Rusiecki JA, Chen L, Srikantan V, Zhang L, Yan L, Polin ML, Baccarelli A (February 2012). "DNA methylation in repetitive elements and post-traumatic stress disorder: a case-control study of US military service members". primary. Epigenomics. 4 (1): 29–40. doi:10.2217/epi.11.116. PMC 3809831. PMID 22332656.
- Chang SC, Koenen KC, Galea S, Aiello AE, Soliven R, Wildman DE, Uddin M (2012). "Molecular variation at the SLC6A3 locus predicts lifetime risk of PTSD in the Detroit Neighborhood Health Study". primary. PLOS ONE. 7 (6): e39184. Bibcode:2012PLoSO...739184C. doi:10.1371/journal.pone.0039184. PMC 3383758. PMID 22745713.
- Rusiecki JA, Byrne C, Galdzicki Z, Srikantan V, Chen L, Poulin M, Yan L, Baccarelli A (2013). "PTSD and DNA Methylation in Select Immune Function Gene Promoter Regions: A Repeated Measures Case-Control Study of U.S. Military Service Members". primary. Frontiers in Psychiatry. 4: 56. doi:10.3389/fpsyt.2013.00056. PMC 3690381. PMID 23805108.
- Labonté B, Azoulay N, Yerko V, Turecki G, Brunet A (March 2014). "Epigenetic modulation of glucocorticoid receptors in posttraumatic stress disorder". primary. Translational Psychiatry. 4 (3): e368. doi:10.1038/tp.2014.3. PMC 3966043. PMID 24594779.
- Yehuda R, Flory JD, Bierer LM, Henn-Haase C, Lehrner A, Desarnaud F, Makotkine I, Daskalakis NP, Marmar CR, Meaney MJ (February 2015). "Lower methylation of glucocorticoid receptor gene promoter 1F in peripheral blood of veterans with posttraumatic stress disorder". primary. Biological Psychiatry. 77 (4): 356–64. doi:10.1016/j.biopsych.2014.02.006. PMID 24661442. S2CID 38006995.
- Vukojevic V, Kolassa IT, Fastenrath M, Gschwind L, Spalek K, Milnik A, Heck A, Vogler C, Wilker S, Demougin P, Peter F, Atucha E, Stetak A, Roozendaal B, Elbert T, Papassotiropoulos A, de Quervain DJ (July 2014). "Epigenetic modification of the glucocorticoid receptor gene is linked to traumatic memory and post-traumatic stress disorder risk in genocide survivors". primary. The Journal of Neuroscience. 34 (31): 10274–84. doi:10.1523/JNEUROSCI.1526-14.2014. PMC 6608273. PMID 25080589.
- Blacker, Caren J.; Frye, Mark A.; Morava, Eva; Kozicz, Tamas; Veldic, Marin (February 13, 2019). "A Review of Epigenetics of PTSD in Comorbid Psychiatric Conditions". Genes. 10 (2): 140. doi:10.3390/genes10020140. ISSN 2073-4425. PMC 6410143. PMID 30781888.
- Dunlop, Boadie W.; Wong, Andrea (2019-03-08). "The hypothalamic-pituitary-adrenal axis in PTSD: Pathophysiology and treatment interventions". Progress in Neuro-Psychopharmacology and Biological Psychiatry. 89: 361–379. doi:10.1016/j.pnpbp.2018.10.010. ISSN 0278-5846. PMID 30342071. S2CID 53044044.
- Uddin M, Aiello AE, Wildman DE, Koenen KC, Pawelec G, de Los Santos R, Goldmann E, Galea S (May 2010). "Epigenetic and immune function profiles associated with posttraumatic stress disorder". primary. Proceedings of the National Academy of Sciences of the United States of America. 107 (20): 9470–5. Bibcode:2010PNAS..107.9470U. doi:10.1073/pnas.0910794107. PMC 2889041. PMID 20439746.
- Blouin, Ashley M.; Sillivan, Stephanie E.; Joseph, Nadine F.; Miller, Courtney A. (October 2016). "The potential of epigenetics in stress-enhanced fear learning models of PTSD". Learning & Memory. 23 (10): 576–586. doi:10.1101/lm.040485.115. ISSN 1072-0502. PMC 5026205. PMID 27634148.
- Yehuda R, Daskalakis NP, Desarnaud F, Makotkine I, Lehrner AL, Koch E, Flory JD, Buxbaum JD, Meaney MJ, Bierer LM (2013). "Epigenetic Biomarkers as Predictors and Correlates of Symptom Improvement Following Psychotherapy in Combat Veterans with PTSD". primary. Frontiers in Psychiatry. 4: 118. doi:10.3389/fpsyt.2013.00118. PMC 3784793. PMID 24098286.
- Zannas AS, Binder EB (January 2014). "Gene-environment interactions at the FKBP5 locus: sensitive periods, mechanisms and pleiotropism". review. Genes, Brain and Behavior. 13 (1): 25–37. doi:10.1111/gbb.12104. PMID 24219237. S2CID 41781060.
- Zhang X, Clark AF, Yorio T (March 2008). "FK506-binding protein 51 regulates nuclear transport of the glucocorticoid receptor beta and glucocorticoid responsiveness". Investigative Ophthalmology & Visual Science. 49 (3): 1037–47. doi:10.1167/iovs.07-1279. PMC 2442563. PMID 18326728.
- Wochnik GM, Rüegg J, Abel GA, Schmidt U, Holsboer F, Rein T (February 2005). "FK506-binding proteins 51 and 52 differentially regulate dynein interaction and nuclear translocation of the glucocorticoid receptor in mammalian cells". primary. The Journal of Biological Chemistry. 280 (6): 4609–16. doi:10.1074/jbc.M407498200. PMID 15591061.
- Xie P, Kranzler HR, Poling J, Stein MB, Anton RF, Farrer LA, Gelernter J (July 2010). "Interaction of FKBP5 with childhood adversity on risk for post-traumatic stress disorder". primary. Neuropsychopharmacology. 35 (8): 1684–92. doi:10.1038/npp.2010.37. PMC 2946626. PMID 20393453.
- Binder EB, Bradley RG, Liu W, Epstein MP, Deveau TC, Mercer KB, Tang Y, Gillespie CF, Heim CM, Nemeroff CB, Schwartz AC, Cubells JF, Ressler KJ (March 2008). "Association of FKBP5 polymorphisms and childhood abuse with risk of posttraumatic stress disorder symptoms in adults". primary. JAMA. 299 (11): 1291–305. doi:10.1001/jama.299.11.1291. PMC 2441757. PMID 18349090.
- Boscarino JA, Erlich PM, Hoffman SN, Zhang X (2012). "Higher FKBP5, COMT, CHRNA5, and CRHR1 allele burdens are associated with PTSD and interact with trauma exposure: implications for neuropsychiatric research and treatment". primary. Neuropsychiatric Disease and Treatment. 8: 131–9. doi:10.2147/NDT.S29508. PMC 3333786. PMID 22536069.
- Klengel T, Mehta D, Anacker C, Rex-Haffner M, Pruessner JC, Pariante CM, Pace TW, Mercer KB, Mayberg HS, Bradley B, Nemeroff CB, Holsboer F, Heim CM, Ressler KJ, Rein T, Binder EB (January 2013). "Allele-specific FKBP5 DNA demethylation mediates gene-childhood trauma interactions". primary. Nature Neuroscience. 16 (1): 33–41. doi:10.1038/nn.3275. PMC 4136922. PMID 23201972.
- Palmisano M, Pandey SC (May 2017). "Epigenetic mechanisms of alcoholism and stress-related disorders". review. Alcohol. 60: 7–18. doi:10.1016/j.alcohol.2017.01.001. PMC 5464725. PMID 28477725.
- Agudelo M, Figueroa G, Parira T, Yndart A, Muñoz K, Atluri V, Samikkannu T, Nair MP (2016). "Profile of Class I Histone Deacetylases (HDAC) by Human Dendritic Cells after Alcohol Consumption and In Vitro Alcohol Treatment and Their Implication in Oxidative Stress: Role of HDAC Inhibitors Trichostatin A and Mocetinostat". primary. PLOS ONE. 11 (6): e0156421. Bibcode:2016PLoSO..1156421A. doi:10.1371/journal.pone.0156421. PMC 4889108. PMID 27249803.
- Moonat S, Sakharkar AJ, Zhang H, Tang L, Pandey SC (April 2013). "Aberrant histone deacetylase2-mediated histone modifications and synaptic plasticity in the amygdala predisposes to anxiety and alcoholism". primary. Biological Psychiatry. 73 (8): 763–73. doi:10.1016/j.biopsych.2013.01.012. PMC 3718567. PMID 23485013.
- Moonat S, Pandey SC (2012). "Stress, epigenetics, and alcoholism". review. Alcohol Research. 34 (4): 495–505. PMC 3860391. PMID 23584115.
- Pandey SC, Ugale R, Zhang H, Tang L, Prakash A (April 2008). "Brain chromatin remodeling: a novel mechanism of alcoholism". primary. The Journal of Neuroscience. 28 (14): 3729–37. doi:10.1523/JNEUROSCI.5731-07.2008. PMC 6671100. PMID 18385331.
- Maron, Eduard; Nutt, David (September 2015). "Biological predictors of pharmacological therapy in anxiety disorders". Dialogues in Clinical Neuroscience. 17 (3): 305–17. doi:10.31887/DCNS.2015.17.3/emaron. PMC 4610615. PMID 26487811.
- Roy-Byrne, Peter (June 2015). "Treatment-refractory anxiety; definition, risk factors, and treatment challenges". Dialogues in Clinical Neuroscience. 17 (2): 191–206. doi:10.31887/DCNS.2015.17.2/proybyrne. PMC 4518702. PMID 26246793.
- Peedicayil, J (March 2020). "The Potential Role of Epigenetic Drugs in the Treatment of Anxiety Disorders". Neuropsychiatric Disease and Treatment. 2020 (16): 597–606. doi:10.2147/NDT.S242040. PMC 7060022. PMID 32184601.
- Eckschlager, Tomas; Plch, Johana; Marie, Stiborova; Hrabeta, Jan (July 2017). "Histone Deacetylase Inhibitors as Anticancer Drugs". International Journal of Molecular Sciences. 18 (7): 1414. doi:10.3390/ijms18071414. PMC 5535906. PMID 28671573.
- Anubama, Rajan; Shi, Hang; Xue, Bingzhong (August 2018). "Class I and II Histone Deacetylase Inhibitors Differentially Regulate Thermogenic Gene Expression in Brown Adipocytes". Scientific Reports. 8 (13072): 13072. Bibcode:2018NatSR...813072R. doi:10.1038/s41598-018-31560-w. PMC 6117331. PMID 30166563.
- Wei, Ya Bin; Melas, Philippe A; Wegener, Gregers; Mathé, Aleksander A.; Lavebratt, Catharina (February 2015). "Antidepressant-Like Effect of Sodium Butyrate is Associated with an Increase in TET1 and in 5-Hydroxymethylation Levels in the Bdnf Gene". International Journal of Neuropsychopharmacology. 18 (2): pyu032. doi:10.1093/ijnp/pyu032. PMC 4368891. PMID 25618518.
- Primeau, Francois; Fontaine, Rejean; Beauclair, Linda (April 1990). "Valproic Acid and Panic Disorder". The Canadian Journal of Psychiatry. 35 (3): 248–250. doi:10.1177/070674379003500309. PMID 2111204. S2CID 37695090.
- Woodman, CL; Noyes Jr., R (April 1994). "Panic disorder: treatment with valproate". The Journal of Clinical Psychiatry. 55 (4): 134–6. PMID 8071256.
- Keck Jr., Paul E.; Taylor, Valerie E.; Tugrul, Karen C.; McElroy, Susan L.; Bennett, Jerry A. (April 1993). "Valproate treatment of panic disorder and lactate-induced panic attacks". Biological Psychiatry. 33 (7): 542–546. doi:10.1016/0006-3223(93)90010-B. PMID 8513040. S2CID 1411404.
- Kinrys, G; Pollack, MH; Simon, NM; Worthington, JJ; Nardi, AE (May 2003). "Valproic acid for the treatment of social anxiety disorder". International Clinical Psychopharmacology. 18 (3): 169–172. doi:10.1097/01.yic.0000064261.66765.9f. PMID 12702897. S2CID 43035988.
- Aliyev, NA; Aliyev, ZN (April 2020). "Valproate (depakine-chrono) in the acute treatment of outpatients with generalized anxiety disorder without psychiatric comorbidity: Randomized, double-blind placebo-controlled study". European Psychiatry. 23 (2): 109–114. doi:10.1016/j.eurpsy.2007.08.001. PMID 17945470. S2CID 20266182.
- Bach, D; Korn, C; Vunder, J; Bantel, A (August 2018). "Effect of valproate and pregabalin on human anxiety-like behaviour in a randomised controlled trial". Translational Psychology. 8 (157): 157. doi:10.1038/s41398-018-0206-7. PMC 6095858. PMID 30115911.
- Whittle, N; Schmuckermair, C; Gunduz Cinar, O; Hauschild, M; Ferraguti, F; Holmes, A; Singewald, Nicolas (January 2013). "Deep brain stimulation, histone deacetylase inhibitors and glutamatergic drugs rescue resistance to fear extinction in a genetic mouse model". Neuropharmacology. 64 (4): 414–423. doi:10.1016/j.neuropharm.2012.06.001. PMC 3474950. PMID 22722028.
- You, C; Zhang, H; Sakharkar, A; Teppen, T; Pandey, S (February 2014). "Reversal of deficits in dendritic spines, BDNF and Arc expression in the amygdala during alcohol dependence by HDAC inhibitor treatment". International Journal of Neuropsychopharmacology. 17 (2): 313–322. doi:10.1017/S1461145713001144. PMC 4093912. PMID 24103311.
- Moloney, RD; Stilling, RM; Dinan, TG; Cryan, JF (September 2015). "Early‐life stress‐induced visceral hypersensitivity and anxiety behavior is reversed by histone deacetylase inhibition". Neurogastroenterology & Motility. 27 (12): 1831–1836. doi:10.1111/nmo.12675. PMID 26403543. S2CID 30923705.
- Hayase, T (July 2016). "Putative Epigenetic Involvement of the Endocannabinoid System in Anxiety- and Depression-Related Behaviors Caused by Nicotine as a Stressor". PLOS ONE. 11 (7): e0158950. Bibcode:2016PLoSO..1158950H. doi:10.1371/journal.pone.0158950. PMC 4942073. PMID 27404492.
- Karnib, N; Sleiman, S; El-Ghandur, R; El Hayek, L; Nasrallah, P; Khalifeh, M; Barmo, N; Jabre, V; Ibrahim, P; Bilen, M; Stephan, J; Holson, E; Ratan, R (January 2019). "Lactate is an antidepressant that mediates resilience to stress by modulating the hippocampal levels and activity of histone deacetylases". Neuropsychopharmacology. 44 (6): 1152–1162. doi:10.1038/s41386-019-0313-z. PMC 6461925. PMID 30647450.
- Kothgassner, Oswald D.; Pellegrini, Marie; Goreis, Andreas; Giordano, Vito; Edobor, Joy; Fischer, Susanne; Plener, Paul L.; Huscsava, Mercedes M. (2021-04-01). "Hydrocortisone administration for reducing post-traumatic stress symptoms: A systematic review and meta-analysis". Psychoneuroendocrinology. 126: 105168. doi:10.1016/j.psyneuen.2021.105168. ISSN 0306-4530. PMID 33626392. S2CID 231930081.
- Zohar, Joseph; Yahalom, Hila; Kozlovsky, Nitsan; Cwikel-Hamzany, Shlomit; Matar, Michael A.; Kaplan, Zeev; Yehuda, Rachel; Cohen, Hagit (2011-11-01). "High dose hydrocortisone immediately after trauma may alter the trajectory of PTSD: Interplay between clinical and animal studies". European Neuropsychopharmacology. 21 (11): 796–809. doi:10.1016/j.euroneuro.2011.06.001. ISSN 0924-977X. PMID 21741804. S2CID 32595065.
- Al Jowf, Ghazi I.; Snijders, Clara; Rutten, Bart P. F.; de Nijs, Laurence; Eijssen, Lars M. T. (October 4, 2021). "The Molecular Biology of Susceptibility to Post-Traumatic Stress Disorder: Highlights of Epigenetics and Epigenomics". International Journal of Molecular Sciences. 22 (19): 10743. doi:10.3390/ijms221910743. ISSN 1422-0067. PMC 8509551. PMID 34639084.
- Youssef, Nagy A. (2022-03-31). "Potential Societal and Cultural Implications of Transgenerational Epigenetic Methylation of Trauma and PTSD: Pathology or Resilience?". The Yale Journal of Biology and Medicine. 95 (1): 171–174. ISSN 0044-0086. PMC 8961703. PMID 35370497.
- Tortella-Feliu, Miquel; Fullana, Miquel A.; Pérez-Vigil, Ana; Torres, Xavier; Chamorro, Jacobo; Littarelli, Sergio A.; Solanes, Aleix; Ramella-Cravaro, Valentina; Vilar, Ana; González-Parra, José A.; Andero, Raül (2019-12-01). "Risk factors for posttraumatic stress disorder: An umbrella review of systematic reviews and meta-analyses". Neuroscience & Biobehavioral Reviews. 107: 154–165. doi:10.1016/j.neubiorev.2019.09.013. ISSN 0149-7634. PMID 31520677. S2CID 202558369.
Further reading
- Schiele MA, Domschke K (March 2018). "Epigenetics at the crossroads between genes, environment and resilience in anxiety disorders". review. Genes, Brain and Behavior. 17 (3): e12423. doi:10.1111/gbb.12423. PMID 28873274.
- Bartlett AA, Singh R, Hunter RG (2017). "Anxiety and Epigenetics". Neuroepigenomics in Aging and Disease. review. Advances in Experimental Medicine and Biology. Vol. 978. pp. 145–166. doi:10.1007/978-3-319-53889-1_8. ISBN 978-3-319-53888-4. PMID 28523545.
- Daniel P. Keating (2017). Born Anxious: The Lifelong Impact of Early Life Adversity - and How to Break the Cycle. St. Martin's Press. ISBN 978-1250075048.
- Nieto SJ, Patriquin MA, Nielsen DA, Kosten TA (2016). "Don't worry; be informed about the epigenetics of anxiety". review. Pharmacology Biochemistry and Behavior. 146–147: 60–72. doi:10.1016/j.pbb.2016.05.006. PMC 4939112. PMID 27189589.
- Hing B, Gardner C, Potash JB (October 2014). "Effects of negative stressors on DNA methylation in the brain: implications for mood and anxiety disorders". review. American Journal of Medical Genetics. Part B, Neuropsychiatric Genetics. 165B (7): 541–54. doi:10.1002/ajmg.b.32265. PMC 5096645. PMID 25139739.
- Hunter RG, McEwen BS (April 2013). "Stress and anxiety across the lifespan: structural plasticity and epigenetic regulation". review. Epigenomics. 5 (2): 177–94. doi:10.2217/epi.13.8. PMID 23566095.